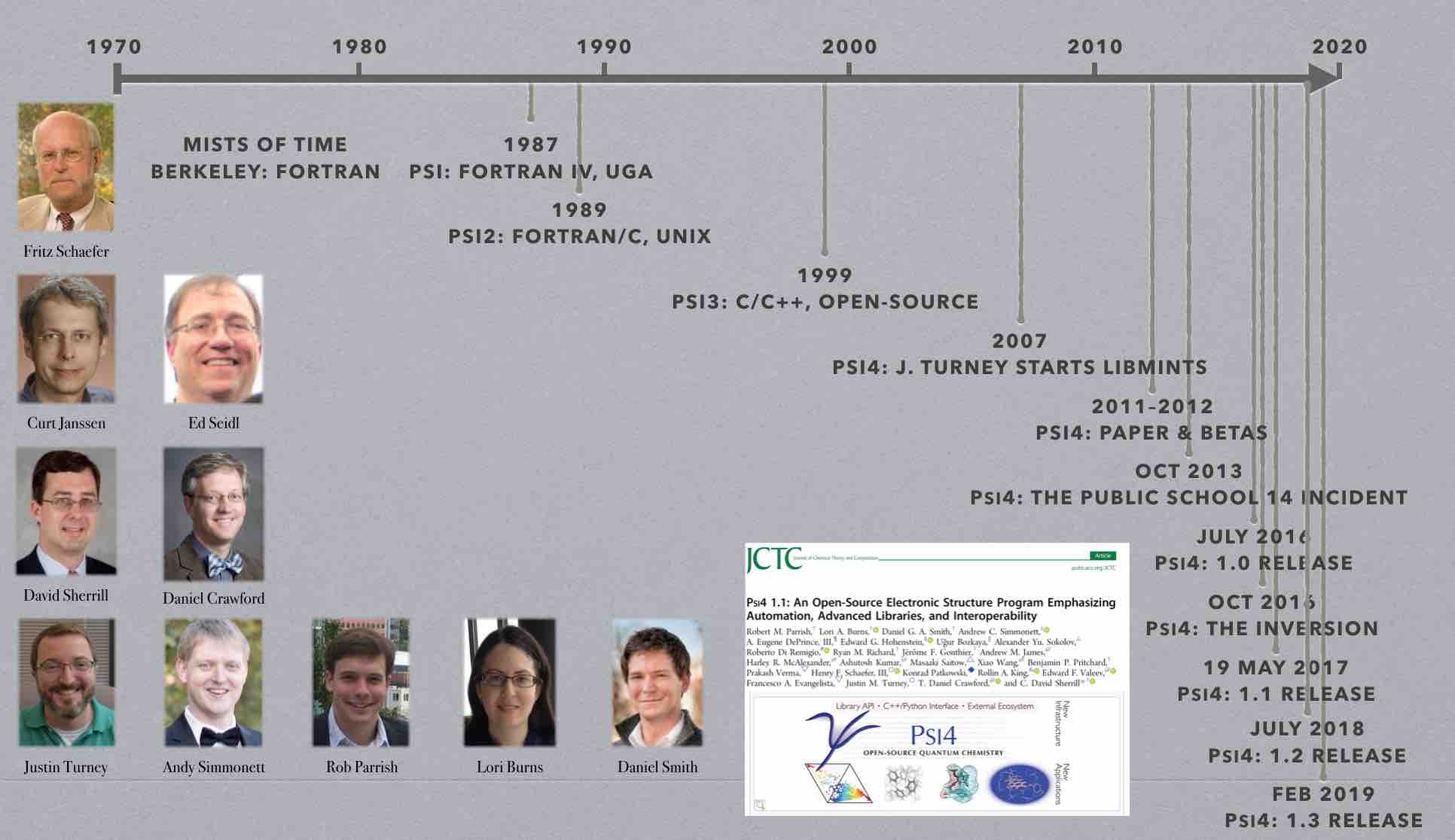
The Psi package originated in the group of Prof. Henry F. Schaefer while
he was at the University of California, Berkeley. The code featured a
direct CI code (written in 1976 by Robert Lucchese), a graphical unitary
group configuration interaction (GUGA-CI) code (written in 1977-79 by
Bernard Brooks and later extended to the shape-driven GUGA-CI code by Paul
Saxe), and a Gaussian integral code (written in 1978 by Russell Pitzer).
The code was originally known as the BERKELEY suite; it was renamed to
Psi after the Schaefer group moved to the University of Georgia. Curt
Janssen and Ed Seidl ported the package to UNIX and started converting it
from Fortran to C. Psi2 featured coupled-cluster codes developed by Curt
Janssen, Gustavo Scuseria, and others, and the INTDER vibrational
analysis and derivative transformation program by Wesley Allen.
Development of Psi3 began in 1999. Much of the infrastructure code was
rewritten to be more flexible on different architectures and to remove
hardwired limits on the number of atoms, basis functions, file sizes, etc.
The new code, developed primarily by Daniel Crawford, David Sherrill, and
Edward Valeev, featured rewritten coupled-cluster code (including
excited-state capabilities), very efficient determinant-based CI code, and
multi-configurational self-consistent-field capabilities. Later releases
of Psi3 added the first production-level implementation of Mukherjee’s
multi-reference coupled-cluster method (PSIMRCC), thanks to Francesco
Evangelista and Andy Simmonett. Psi3 was released free of charge under an
open-source license.
Development of PSI4 began around 2007. PSI4 introduces a completely new input parser and driver program which were designed with two goals in mind: (1) to make the input as user-friendly as possible, and (2) to make complex tasks very easy to perform. This work was performed primarily by Andy Simmonett, Justin Turney, and Lori Burns. Standard computations are specified by a very simple syntax, but the user is also free to add Python code to the input file in order to automate more complex procedures (such as potential energy scans). Built-in Python subroutines handle procedures like counterpoise correction and basis set extrapolation. A new database functionality, developed by Lori Burns, allows the program to automatically perform a series of computations on all members in a given collection of molecules (and tabulate errors against benchmark energies in the database, if desired). A PSI4 interface to the WebMO graphical user interface, released in 2013, was developed by Ashley Ringer McDonald and Matthew Kennedy, with contributions from Eugene DePrince.
A new layer of utility functions was developed by Justin Turney in 2009-12
to make it easier for developers to implement new functionality in PSI4.
Especially important is the LIBMINTS library which makes it easier to
access one- and two-electron integrals from the underlying LIBINT
library of Edward Valeev (the LIBINT library is based on work in the
late 1990’s by Justin Fermann and Edward Valeev). Following up on earlier
work and suggestions by Edward Valeev that we migrate Psi to an
object-oriented C++ design, Justin Turney also developed various C++
classes for basis sets, molecules, and wavefunctions, inspired by similar
work done much earlier by the Massively Parallel Quantum Chemistry
(MPQC) package; work on these classes is ongoing. To ease the path
toward adding distributed-parallel capabilities, PSI4 moved the code to a
“single-executable” model, away from the previous model in which each
program module (integrals, SCF, etc.) was a separate executable program,
with information passed by files.
Other new features developed for PSI4 include a new “plugin” system, written by Justin Turney and Andy Simmonett, that allows developers to create a new feature for PSI4 in a standalone module that can be compiled separately and loaded at runtime. This helps decouple development of the core PSI4 capabilities from the development of new features, shortening the development cycle. Edward Hohenstein wrote a new, efficient density-fitted symmetry-adapted perturbation theory (SAPT) code for PSI4 in 2010-11. A newer SAPT code was developed by Rob Parrish in 2014-15 to allow atomic- and fragment-based partitioning of the energies (A-SAPT and F-SAPT) and intramolecular SAPT (I-SAPT). Rob also brought density functional theory (DFT) capabilities to PSI4 in 2011-12. The DFT framework allows any functional to be entered into MATLAB, and using automatic-differentiation techniques, C++ code is automatically generated. This allows PSI4 to include essentially all popular density functionals. A new density-fitted/Cholesky decomposition (DF/CD) coupled-cluster code for CCSD(T) energies was added by Eugene DePrince in 2013. The DF/CD approximations work very well in concert with Frozen Natural Orbitals (FNO’s), a capability also released in 2013.
In 2012-14, Alex Sokolov added several variants of density cumulant functional theory to PSI4 . Ugur Bozkaya added several analytic gradient methods from 2012-15, including MP2, MP3, MP2.5, CEPA(0), and optimized orbital versions of all these. Density-fitted versions of MP2 (including UHF references) and optimized-orbital MP2 analytic gradients were also added. In 2015, Dr. Bozkaya added density-fitted CCSD analytic gradients.
Lori Burns introduced numerous major generalizations of the PSI4
driver
from 2014-15, allowing it to call features from other programs like
CFOUR and MOLPRO and perform basic combinations of these results with
PSI4
results. This formed the basis of our current project to develop a
Common Driver for Quantum Chemistry. In early 2016, Daniel Smith and Lori
Burns changed the way information is passed from one module to another in
PSI4
, using the Wavefunction object. This new mechanism allowed orbitals
to be passed between modules in a more flexible way, and it allowed
operations like exporting natural orbitals to Molden for plotting, etc.
Interfaces to additional external projects like libefp, GDMA, ChemPS2, and
PCMSolver were added or extended. With these additions, the longstanding
“Beta” tag was removed and PSI4
1.0 was released in July 2016, with both
source code and pre-packaged binaries available for Linux and Mac.
Substantial additional changes to the driver were made in 2016 by Lori Burns and Daniel Smith, allowing us to make PSI4 a full-fledged Python library (wrapping a C++ core). This allows users to call PSI4 from a Python script, and greatly simplifies interoperability with other Python modules. Also in 2016, the code attained analytic RHF CCSD(T) gradients, analytic RHF Hessians, and fragment-based, intramolecular, and open-shell SAPT. Support for Grimme’s semi-empirical HF-3C and PBEh-3C was also added. The 1.1 release was announced at the 2016 SETCA meeting, and an accompanying paper was published by J. Chem. Theory Comput.
Psi Publications
Cumulative Psi Author List
- Robert M. Parrish, Center for Computational Molecular Science and Technology, Georgia Institute of Technology
- Lori A. Burns, Center for Computational Molecular Science and Technology, Georgia Institute of Technology
- Daniel G. A. Smith, The Molecular Sciences Software Institute
- Andrew C. Simmonett, National Institutes of Health -- National Heart, Lung and Blood Institute
- A. Eugene DePrince, Department of Chemistry and Biochemistry, Florida State University
- Edward G. Hohenstein, PULSE Institute for Ultrafast Energy Sciences, SLAC National Accelerator Laboratory
- Uȣur Bozkaya, Department of Chemistry, Hacettepe University, Turkey
- Alexander Yu. Sokolov, Department of Chemistry and Biochemistry, The Ohio State University
- Roberto Di Remigio, Hylleraas Centre for Quantum Molecular Sciences, UiT - The Arctic University of Norway
- Ryan M. Richard, Center for Computational Molecular Science and Technology, Georgia Institute of Technology
- Jũrƀme F. Gonthier, Center for Computational Molecular Science and Technology, Georgia Institute of Technology
- Andrew M. James, Department of Chemistry, Virginia Tech
- Harley R. McAlexander, Department of Chemistry, Virginia Tech
- Ashutosh Kumar, Department of Chemistry, Virginia Tech
- Masaaki Saitow, Department of Chemistry and Research Center for Smart Molecules, Rikkyo University, Japan
- Xiao Wang, Department of Chemistry, Virginia Tech
- Benjamin P. Pritchard, The Molecular Sciences Software Institute
- Prakash Verma, Department of Chemistry, Emory University
- Henry F. Schaefer, Center for Computational Quantum Chemistry, University of Georgia
- Konrad Patkowski, Department of Chemistry and Biochemistry, Auburn University
- Rollin A. King, Department of Chemistry, Bethel University
- Edward F. Valeev, Department of Chemistry, Virginia Tech
- Francesco A. Evangelista, Department of Chemistry, Emory University
- Justin M. Turney, Center for Computational Quantum Chemistry, University of Georgia
- T. Daniel Crawford, Department of Chemistry, Virginia Tech
- C. David Sherrill, Center for Computational Molecular Science and Technology, Georgia Institute of Technology
- Peter Kraus, School of Molecular and Life Sciences, Curtin University
- Holger Kruse, Institute of Biophysics of the CAS, v.v.i., Czech Republic
- Marvin H. Lechner, Department of Chemistry, Technical University of Munich
- Matthew C. Schieber, Center for Computational Molecular Science and Technology, Georgia Institute of Technology
- Robert A. Shaw, University of Sheffield
- Edward T. Seidl, Center for Computational Quantum Chemistry, University of Georgia
- Curtis L. Janssen, Center for Computational Quantum Chemistry, University of Georgia
- Matthew L. Leininger, Center for Computational Molecular Science and Technology, Georgia Institute of Technology
- Asem Alenaizan, Chemistry Department, King Fahd University of Petroleum and Minerals
- Raimondas Galvelis, Acellera
- Zachary L. Glick, Center for Computational Molecular Science and Technology, Georgia Institute of Technology
- Susi Lehtola, Department of Chemistry, University of Helsinki
- Jonathon P. Misiewicz, Department of Chemistry, Emory University
- Maximilian Scheurer, Interdisciplinary Center for Scientific Computing, Heidelberg University
- Jeffrey B. Schriber, Center for Computational Molecular Science and Technology, Georgia Institute of Technology
- Yi Xie, Center for Computational Molecular Science and Technology, Georgia Institute of Technology
- Dominic A. Sirianni, Center for Computational Molecular Science and Technology, Georgia Institute of Technology
- Joseph Senan O'Brien, Center for Computational Molecular Science and Technology, Georgia Institute of Technology
- Jonathan M. Waldrop, Department of Chemistry and Biochemistry, Auburn University
- Andy Jiang, Center for Computational Molecular Science and Technology, Georgia Institute of Technology
- Stefan Behnle, Eberhard Karls Universität Tübingen Mathematisch-Naturwissenschaftliche Fakultät
- David L. Poole, Center for Computational Molecular Science and Technology, Georgia Institute of Technology
- Alexander G. Heide, Center for Computational Quantum Chemistry, University of Georgia
- Tibor Győri, Department of Physical Chemistry and Materials Science, University of Szeged
- Erica C. Mitchell, Center for Computational Quantum Chemistry, University of Georgia
- John P. Pederson, School of Chemistry and Biochemistry, Georgia Institute of Technology
- Austin M. Wallace, Center for Computational Molecular Science and Technology, Georgia Institute of Technology