
SCF¶
Performs self consistent field (Hartree-Fock and Density Functional Theory) computations. These are the starting points for most computations, so this code is called in most cases.
General Wavefunction Info¶
BASIS¶
Primary basis set
- Type: string
- Possible Values: basis string
- Default: No Default
DF_BASIS_SCF¶
Auxiliary basis set for SCF density fitting computations. Defaults to a JKFIT basis.
- Type: string
- Possible Values: basis string
- Default: No Default
DF_SCF_GUESS¶
Use DF integrals tech to converge the SCF before switching to a conventional tech
- Type: boolean
- Default: true
GUESS¶
The type of guess orbitals. Defaults to CORE except for geometry optimizations, in which case READ becomes the default after the first geometry step.
- Type: string
- Possible Values: CORE, GWH, SAD, READ
- Default: CORE
INTS_TOLERANCE¶
Minimum absolute value below which TEI are neglected.
- Type: conv double
- Default: 0.0
MOLDEN_WRITE¶
Do write a MOLDEN output file? If so, the filename will end in .molden, and the prefix is determined by WRITER_FILE_LABEL (if set), or else by the name of the output file plus the name of the current molecule.
- Type: boolean
- Default: false
PRINT_BASIS¶
Flag to print the basis set.
- Type: boolean
- Default: false
REFERENCE¶
Reference wavefunction type
- Type: string
- Possible Values: RHF, ROHF, UHF, CUHF, RKS, UKS
- Default: RHF
SCF_MEM_SAFETY_FACTOR¶
Memory safety factor for allocating JK
- Type: double
- Default: 0.75
SCF_TYPE¶
What algorithm to use for the SCF computation. See Table SCF Convergence & Algorithm for default algorithm for different calculation types.
- Type: string
- Possible Values: DIRECT, DF, PK, OUT_OF_CORE
- Default: PK
S_ORTHOGONALIZATION¶
SO orthogonalization: symmetric or canonical?
- Type: string
- Possible Values: SYMMETRIC, CANONICAL
- Default: SYMMETRIC
S_TOLERANCE¶
Minimum S matrix eigenvalue to be used before compensating for linear dependencies.
- Type: conv double
- Default: 1e-7
Convergence Control/Stabilization¶
BASIS_GUESS¶
Accelerate convergence by performing a preliminary scf with this small basis set followed by projection into the full target basis. A value of TRUE turns on projection using the 3-21G small basis set.
- Type: string
- Default: FALSE
DAMPING_CONVERGENCE¶
The density convergence threshold after which damping is no longer performed, if it is enabled. It is recommended to leave damping on until convergence, which is the default.
- Type: conv double
- Default: 1.0e-18
DAMPING_PERCENTAGE¶
The amount (percentage) of damping to apply to the early density updates. 0 will result in a full update, 100 will completely stall the update. A value around 20 (which corresponds to 20% of the previous iteration’s density being mixed into the current density) could help to solve problems with oscillatory convergence.
- Type: double
- Default: 100.0
DF_BASIS_GUESS¶
When BASIS_GUESS is active, run the preliminary scf in density-fitted mode with this as fitting basis for the small basis set. A value of TRUE turns on density fitting with the cc-pVDZ-RI basis set (when available for all elements).
- Type: string
- Possible Values: basis string
- Default: FALSE
DIIS_MAX_VECS¶
Maximum number of error vectors stored for DIIS extrapolation
- Type: integer
- Default: 10
DIIS_MIN_VECS¶
Minimum number of error vectors stored for DIIS extrapolation
- Type: integer
- Default: 2
DIIS_START¶
The minimum iteration to start storing DIIS vectors
- Type: integer
- Default: 1
D_CONVERGENCE¶
Convergence criterion for SCF density, which is defined as the RMS value of the orbital gradient. See Table SCF Convergence & Algorithm for default convergence criteria for different calculation types.
- Type: conv double
- Default: 1e-8
E_CONVERGENCE¶
Convergence criterion for SCF energy. See Table SCF Convergence & Algorithm for default convergence criteria for different calculation types.
- Type: conv double
- Default: 1e-8
FAIL_ON_MAXITER¶
Fail if we reach maxiter without converging?
- Type: boolean
- Default: true
MOM_OCC¶
The absolute indices of orbitals to excite from in MOM (+/- for alpha/beta)
- Type: array
- Default: No Default
MOM_VIR¶
The absolute indices of orbitals to excite to in MOM (+/- for alpha/beta)
- Type: array
- Default: No Default
STABILITY_ANALYSIS¶
Whether to perform stability analysis after convergence. NONE prevents analysis being performed. CHECK will print out the analysis of the wavefunction stability at the end of the computation. FOLLOW will perform the analysis and, if a totally symmetric instability is found, will attemp to follow the eigenvector and re-run the computations to find a stable solution.
- Type: string
- Possible Values: NONE, CHECK, FOLLOW
- Default: NONE
Fractional Occupation UHF/UKS¶
FRAC_OCC¶
The absolute indices of occupied orbitals to fractionally occupy (+/- for alpha/beta)
- Type: array
- Default: No Default
FRAC_RENORMALIZE¶
Do renormalize C matrices prior to writing to checkpoint?
- Type: boolean
- Default: true
FRAC_START¶
The iteration to start fractionally occupying orbitals (or 0 for no fractional occupation)
- Type: integer
- Default: 0
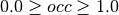 )
)Environmental Effects¶
ONEPOT_GRID_READ¶
Read an external potential from the .dx file?
- Type: boolean
- Default: false
PERTURB_MAGNITUDE¶
Size of the perturbation (applies only to dipole perturbations)
- Type: double
- Default: 0.0
PERTURB_WITH¶
The operator used to perturb the Hamiltonian, if requested
- Type: string
- Possible Values: DIPOLE_X, DIPOLE_Y, DIPOLE_Z, EMBPOT, SPHERE, DX
- Default: DIPOLE_X
PHI_POINTS¶
Number of azimuthal grid points for sphereical potential integration
- Type: integer
- Default: 360
R_POINTS¶
Number of radial grid points for sphereical potential integration
- Type: integer
- Default: 100
THETA_POINTS¶
Number of colatitude grid points for sphereical potential integration
- Type: integer
- Default: 360
DFSCF Algorithm¶
DF_INTS_NUM_THREADS¶
Number of threads for integrals (may be turned down if memory is an issue). 0 is blank
- Type: integer
- Default: 0
SAD Guess Algorithm¶
SAD_D_CONVERGENCE¶
Convergence criterion for SCF density in SAD Guess.
- Type: conv double
- Default: 1e-5
SAD_E_CONVERGENCE¶
Convergence criterion for SCF energy in SAD Guess.
- Type: conv double
- Default: 1e-5
DFT¶
DFT_BASIS_TOLERANCE¶
DFT basis cutoff.
- Type: conv double
- Default: 1.0e-12
DFT_BS_RADIUS_ALPHA¶
Factor for effective BS radius in radial grid.
- Type: double
- Default: 1.0
DFT_CUSTOM_FUNCTIONAL¶
A custom DFT functional object (built by Python or NULL/None)
- Type: python
- Default: No Default
DFT_DISPERSION_PARAMETERS¶
Parameters defining the dispersion correction. See Table -D Functionals for default values and Table Dispersion Corrections for the order in which parameters are to be specified in this array option.
- Type: array
- Default: No Default
DFT_FUNCTIONAL¶
The DFT combined functional name, e.g. B3LYP, or GEN to use a python reference to a custom functional specified by DFT_CUSTOM_FUNCTIONAL.
- Type: string
- Default: No Default
DFT_NUCLEAR_SCHEME¶
Nuclear Scheme.
- Type: string
- Possible Values: TREUTLER, BECKE, NAIVE, STRATMANN
- Default: TREUTLER
DFT_RADIAL_POINTS¶
Number of radial points.
- Type: integer
- Default: 75
DFT_RADIAL_SCHEME¶
Radial Scheme.
- Type: string
- Possible Values: TREUTLER, BECKE, MULTIEXP, EM, MURA
- Default: TREUTLER
DFT_SPHERICAL_POINTS¶
Number of spherical points (A Lebedev Points number).
- Type: integer
- Default: 302
DFT_SPHERICAL_SCHEME¶
Spherical Scheme.
- Type: string
- Possible Values: LEBEDEV
- Default: LEBEDEV
Expert General Wavefunction Info¶
Expert Convergence Control/Stabilization¶
FOLLOW_STEP_SCALE¶
When using STABILITY_ANALYSIS = FOLLOW, how much to scale the step along the eigenvector by.
- Type: double
- Default: 0.5
Expert Parallel Runtime¶
DISTRIBUTED_MATRIX¶
The dimension sizes of the distributed matrix
- Type: array
- Default: No Default
PROCESS_GRID¶
The dimension sizes of the processor grid
- Type: array
- Default: No Default
Expert Misc.¶
Expert DFSCF Algorithm¶
DF_FITTING_CONDITION¶
Fitting Condition
- Type: double
- Default: 1.0e-12
DF_INTS_IO¶
IO caching for CP corrections, etc
- Type: string
- Possible Values: NONE, SAVE, LOAD
- Default: NONE
Expert SAD Guess Algorithm¶
SAD_CHOL_TOLERANCE¶
SAD Guess Cholesky Cutoff (for eliminating redundancies).
- Type: conv double
- Default: 1e-7
SAD_F_MIX_START¶
SAD Guess F-mix Iteration Start
- Type: integer
- Default: 50
SAD_MAXITER¶
Maximum number of SAD guess iterations
- Type: integer
- Default: 50
Expert DFT¶
DFT_BLOCK_MAX_POINTS¶
The maximum number of grid points per evaluation block.
- Type: integer
- Default: 5000
DFT_BLOCK_MAX_RADIUS¶
The maximum radius to terminate subdivision of an octree block [au].
- Type: double
- Default: 3.0
DFT_BLOCK_MIN_POINTS¶
The minimum number of grid points per evaluation block.
- Type: integer
- Default: 1000
DFT_BLOCK_SCHEME¶
The blocking scheme for DFT.
- Type: string
- Possible Values: NAIVE, OCTREE
- Default: OCTREE
DFT_GRID_NAME¶
The DFT grid specification, such as SG1.
- Type: string
- Possible Values: SG1
- Default: No Default
DFT_PRUNING_ALPHA¶
Spread alpha for logarithmic pruning.
- Type: double
- Default: 1.0
DFT_PRUNING_SCHEME¶
Pruning Scheme.
- Type: string
- Possible Values: FLAT, P_GAUSSIAN, D_GAUSSIAN, P_SLATER, D_SLATER, LOG_GAUSSIAN, LOG_SLATER
- Default: FLAT