
Keywords by Alpha¶
- A_RAS3_MAX (DETCI)
DETCI — maximum number of alpha electrons in RAS III
- Type: integer
- Default: -1
- AA_M_FILE (TRANSQT)
TRANSQT — MO basis (PQ|RS) type two-electron integrals file
- Type: integer
- Default: PSIF_MO_AA_TEI
- AB_M_FILE (TRANSQT)
TRANSQT — MO basis (PQ|rs) type two-electron integrals file
- Type: integer
- Default: PSIF_MO_AB_TEI
- ABCD (CCENERGY)
CCENERGY — Type of ABCD algorithm will be used
- Type: string
- Possible Values: NEW, OLD
- Default: NEW
- ABCD (CCEOM)
CCEOM — Type of ABCD algorithm will be used
- Type: string
- Possible Values: NEW, OLD
- Default: NEW
- ABCD (CCLAMBDA)
CCLAMBDA — Type of ABCD algorithm will be used
- Type: string
- Default: NEW
- ABCD (CCRESPONSE)
CCRESPONSE — Type of ABCD algorithm will be used
- Type: string
- Default: NEW
- ACTIVE (DETCI)
DETCI — An array giving the number of active orbitals (occupied plus unoccupied) per irrep (shorthand to make MCSCF easier to specify than using RAS keywords)
- Type: array
- Default: No Default
- ACTIVE (PSIMRCC)
PSIMRCC — The number of active orbitals per irrep
- Type: array
- Default: No Default
- ADD_AUXILIARY_BONDS (OPTKING)
OPTKING — Do add bond coordinates at nearby atoms for non-bonded systems?
- Type: boolean
- Default: false
- AEL (CCDENSITY)
CCDENSITY (Expert) — Do compute the approximate excitation level? See Stanton and Bartlett, JCP, 98, 1993, 7034.
- Type: boolean
- Default: false
- AIO_CPHF (SAPT)
SAPT — Do use asynchronous disk I/O in the solution of the CPHF equations? Use may speed up the computation slightly at the cost of spawning an additional thread.
- Type: boolean
- Default: false
- AIO_DF_INTS (SAPT)
SAPT — Do use asynchronous disk I/O in the formation of the DF integrals? Use may speed up the computation slightly at the cost of spawning an additional thread.
- Type: boolean
- Default: false
- ALGORITHM (DCFT)
DCFT — The algorithm to use for the density cumulant and orbital updates in the DCFT energy computation. Two-step algorithm (default) is usually more efficient for small systems, but for large systems the simultaneous algorithm is recommended. In the cases where the convergence problems are encountered (especially for highly symmetric systems) QC algorithm can be used.
- Type: string
- Possible Values: TWOSTEP, SIMULTANEOUS, QC
- Default: TWOSTEP
- ANALYZE (CCENERGY)
CCENERGY — Do analyze T2 amplitudes
- Type: boolean
- Default: false
- ANALYZE (CCRESPONSE)
CCRESPONSE — Do analyze X2 amplitudes
- Type: boolean
- Default: false
- AO_BASIS (CCDENSITY)
CCDENSITY — The algorithm to use for the
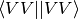 terms
terms- Type: string
- Possible Values: NONE, DISK, DIRECT
- Default: NONE
- AO_BASIS (CCENERGY)
CCENERGY (Expert) — The algorithm to use for the
terms If AO_BASIS is NONE, the MO-basis integrals will be used; if AO_BASIS is DISK, the AO-basis integrals stored on disk will be used; if AO_BASIS is DIRECT, the AO-basis integrals will be computed on the fly as necessary. NB: The DIRECT option is not fully implemented and should only be used by experts. Default is NONE. Note: The developers recommend use of this keyword only as a last resort because it significantly slows the calculation. The current algorithms for handling the MO-basis four-virtual-index integrals have been significantly improved and are preferable to the AO-based approach.- Type: string
- Possible Values: NONE, DISK, DIRECT
- Default: NONE
- AO_BASIS (CCLAMBDA)
CCLAMBDA — The algorithm to use for the
terms- Type: string
- Possible Values: NONE, DISK, DIRECT
- Default: NONE
- AO_BASIS (CCSORT)
CCSORT — The algorithm to use for the
terms- Type: string
- Possible Values: NONE, DISK, DIRECT
- Default: NONE
- AO_BASIS (DCFT)
DCFT — Controls whether to avoid the AO->MO transformation of the two-electron integrals for the four-virtual case (<VV||VV>) by computing the corresponding terms in the AO basis. AO_BASIS = DISK algorithm reduces the memory requirements and can significantly reduce the cost of the energy computation if SIMULTANEOUS algorithm is used. For the TWOSTEP algorithm, however, AO_BASIS = DISK option is not recommended due to the extra I/O.
- Type: string
- Possible Values: NONE, DISK
- Default: NONE
- AO_BASIS (TRANSQT)
TRANSQT — The algorithm to use for the
terms- Type: string
- Possible Values: NONE, DISK, DIRECT
- Default: NONE
- AO_BASIS (TRANSQT2)
TRANSQT2 — The algorithm to use for the
terms- Type: string
- Possible Values: NONE, DISK, DIRECT
- Default: NONE
- AVG_STATES (DETCI)
DETCI — Array giving the root numbers of the states to average in a state-averaged procedure such as SA-CASSCF. Root numbering starts from 1.
- Type: array
- Default: No Default
- AVG_WEIGHTS (DETCI)
DETCI — Array giving the weights for each state in a state-averaged procedure
- Type: array
- Default: No Default
- B_RAS3_MAX (DETCI)
DETCI — maximum number of beta electrons in RAS III
- Type: integer
- Default: -1
- BASIS (DFMP2)
DFMP2 — Primary basis set
- Type: string
- Possible Values: basis string
- Default: NONE
- BASIS (DFTSAPT)
DFTSAPT — The name of the orbital basis set
- Type: string
- Possible Values: basis string
- Default: No Default
- BASIS (MINTS)
MINTS — Primary basis set
- Type: string
- Possible Values: basis string
- Default: No Default
- BASIS (SAPT)
SAPT — Primary basis set, describes the monomer molecular orbitals
- Type: string
- Possible Values: basis string
- Default: No Default
- BASIS (SCF)
SCF — Primary basis set
- Type: string
- Possible Values: basis string
- Default: No Default
- BASIS_GUESS (SCF)
SCF — Accelerate convergence by performing a preliminary scf with this small basis set followed by projection into the full target basis. A value of TRUE turns on projection using the 3-21G small basis set.
- Type: string
- Default: FALSE
- BB_M_FILE (TRANSQT)
TRANSQT — MO basis (pq|rs) type two-electron integrals file
- Type: integer
- Default: PSIF_MO_BB_TEI
- BENCH (DFTSAPT)
DFTSAPT — Bench level
- Type: integer
- Default: 0
- BENCH (GLOBALS)
GLOBALS — Some codes (DFT) can dump benchmarking data to separate output files
- Type: integer
- Default: 0
- BENDAZZOLI (DETCI)
DETCI (Expert) — Do use some routines based on the papers of Bendazzoli et al. to calculate sigma? Seems to be slower and not worthwhile; may disappear eventually. Works only for full CI and I don’t remember if I could see how their clever scheme might be extended to RAS in general.
- Type: boolean
- Default: false
- BRUECKNER_MAXITER (FNOCC)
FNOCC — Maximum number of iterations for Brueckner orbitals optimization
- Type: integer
- Default: 20
- BRUECKNER_ORBS_R_CONVERGENCE (CCENERGY)
CCENERGY — Convergence criterion for Breuckner orbitals. The convergence is determined based on the largest
 amplitude. Default adjusts depending on E_CONVERGENCE.
amplitude. Default adjusts depending on E_CONVERGENCE.- Type: conv double
- Default: 1e-5
- CACHELEVEL (ADC)
ADC — How to cache quantities within the DPD library
- Type: integer
- Default: 2
- CACHELEVEL (CCDENSITY)
CCDENSITY — The amount of cacheing of data to perform
- Type: integer
- Default: 2
- CACHELEVEL (CCENERGY)
CCENERGY — Cacheing level for libdpd governing the storage of amplitudes, integrals, and intermediates in the CC procedure. A value of 0 retains no quantities in cache, while a level of 6 attempts to store all quantities in cache. For particularly large calculations, a value of 0 may help with certain types of memory problems. The default is 2, which means that all four-index quantites with up to two virtual-orbital indices (e.g.,
 integrals) may be held in the cache.
integrals) may be held in the cache.- Type: integer
- Default: 2
- CACHELEVEL (CCEOM)
CCEOM — Cacheing level for libdpd governing the storage of amplitudes, integrals, and intermediates in the CC procedure. A value of 0 retains no quantities in cache, while a level of 6 attempts to store all quantities in cache. For particularly large calculations, a value of 0 may help with certain types of memory problems. The default is 2, which means that all four-index quantites with up to two virtual-orbital indices (e.g.,
integrals) may be held in the cache.- Type: integer
- Default: 2
- CACHELEVEL (CCHBAR)
CCHBAR — Cacheing level for libdpd governing the storage of amplitudes, integrals, and intermediates in the CC procedure. A value of 0 retains no quantities in cache, while a level of 6 attempts to store all quantities in cache. For particularly large calculations, a value of 0 may help with certain types of memory problems. The default is 2, which means that all four-index quantites with up to two virtual-orbital indices (e.g.,
integrals) may be held in the cache.- Type: integer
- Default: 2
- CACHELEVEL (CCLAMBDA)
CCLAMBDA — Cacheing level for libdpd governing the storage of amplitudes, integrals, and intermediates in the CC procedure. A value of 0 retains no quantities in cache, while a level of 6 attempts to store all quantities in cache. For particularly large calculations, a value of 0 may help with certain types of memory problems. The default is 2, which means that all four-index quantites with up to two virtual-orbital indices (e.g.,
integrals) may be held in the cache.- Type: integer
- Default: 2
- CACHELEVEL (CCRESPONSE)
CCRESPONSE — Cacheing level for libdpd
- Type: integer
- Default: 2
- CACHELEVEL (CCSORT)
CCSORT — Cacheing level for libdpd governing the storage of amplitudes, integrals, and intermediates in the CC procedure. A value of 0 retains no quantities in cache, while a level of 6 attempts to store all quantities in cache. For particularly large calculations, a value of 0 may help with certain types of memory problems. The default is 2, which means that all four-index quantites with up to two virtual-orbital indices (e.g.,
integrals) may be held in the cache.- Type: integer
- Default: 2
- CACHELEVEL (DCFT)
DCFT (Expert) — Controls how to cache quantities within the DPD library
- Type: integer
- Default: 2
- CACHELEVEL (OCC)
OCC — Cacheing level for libdpd governing the storage of amplitudes, integrals, and intermediates in the CC procedure. A value of 0 retains no quantities in cache, while a level of 6 attempts to store all quantities in cache. For particularly large calculations, a value of 0 may help with certain types of memory problems. The default is 2, which means that all four-index quantites with up to two virtual-orbital indices (e.g.,
integrals) may be held in the cache.- Type: integer
- Default: 2
- CACHELEVEL (STABILITY)
-
- Type: integer
- Default: 2
- CACHELEVEL (TRANSQT2)
TRANSQT2 (Expert) — Controls how to cache quantities within the DPD library
- Type: integer
- Default: 2
- CACHETYPE (CCENERGY)
CCENERGY — Selects the priority type for maintaining the automatic memory cache used by the libdpd codes. A value of LOW selects a “low priority” scheme in which the deletion of items from the cache is based on pre-programmed priorities. A value of LRU selects a “least recently used” scheme in which the oldest item in the cache will be the first one deleted.
- Type: string
- Possible Values: LOW, LRU
- Default: LOW
- CACHETYPE (CCEOM)
CCEOM — The criterion used to retain/release cached data
- Type: string
- Possible Values: LOW, LRU
- Default: LRU
- CANONICALIZE_ACTIVE_FAVG (MCSCF)
MCSCF — Do canonicalize the active orbitals such that the average Fock matrix is diagonal?
- Type: boolean
- Default: false
- CANONICALIZE_INACTIVE_FAVG (MCSCF)
MCSCF — Do canonicalize the inactive (DOCC and Virtual) orbitals such that the average Fock matrix is diagonal?
- Type: boolean
- Default: false
- CART_HESS_READ (OPTKING)
OPTKING — Do read Cartesian Hessian? Only for experts - use FULL_HESS_EVERY instead.
- Type: boolean
- Default: false
- CAS_FILES_WRITE (CLAG)
CLAG — Do write the OEI, TEI, OPDM, TPDM, and Lagrangian files in canonical form, Pitzer order?
- Type: boolean
- Default: false
- CC (DETCI)
DETCI — Do coupled-cluster computation?
- Type: boolean
- Default: false
- CC3_FOLLOW_ROOT (CCEOM)
CCEOM — Do turn on root following for CC3
- Type: boolean
- Default: false
- CC_A_RAS3_MAX (DETCI)
DETCI — maximum number of alpha electrons in RAS III, for CC
- Type: integer
- Default: -1
- CC_B_RAS3_MAX (DETCI)
DETCI — maximum number of beta electrons in RAS III, for CC
- Type: integer
- Default: -1
- CC_DIIS_MAX_VECS (OCC)
OCC — Maximum number of vectors used in amplitude DIIS
- Type: integer
- Default: 6
- CC_DIIS_MIN_VECS (OCC)
OCC — Minimum number of vectors used in amplitude DIIS
- Type: integer
- Default: 2
- CC_EX_LEVEL (DETCI)
DETCI — The CC excitation level
- Type: integer
- Default: 2
- CC_FIX_EXTERNAL (DETCI)
DETCI (Expert) — Do fix amplitudes involving RAS I or RAS IV? Useful in mixed MP2-CC methods.
- Type: boolean
- Default: false
- CC_FIX_EXTERNAL_MIN (DETCI)
DETCI (Expert) — Number of external indices before amplitude gets fixed by CC_FIX_EXTERNAL. Experimental.
- Type: integer
- Default: 1
- CC_MACRO (DETCI)
DETCI (Expert) — CC_MACRO = [ [ex_lvl, max_holes_I, max_parts_IV, max_I+IV], [ex_lvl, max_holes_I, max_parts_IV, max_I+IV], ... ] Optional additional restrictions on allowed exictations in coupled-cluster computations, based on macroconfiguration selection. For each sub-array, [ex_lvl, max_holes_I, max_parts_IV, max_I+IV], eliminate cluster amplitudes in which: [the excitation level (holes in I + II) is equal to ex_lvl] AND [there are more than max_holes_I holes in RAS I, there are more than max_parts_IV particles in RAS IV, OR there are more than max_I+IV quasiparticles in RAS I + RAS IV].
- Type: array
- Default: No Default
- CC_MAXITER (OCC)
OCC — Maximum number of iterations to determine the amplitudes
- Type: integer
- Default: 50
- CC_MIXED (DETCI)
DETCI (Expert) — Do ignore block if num holes in RAS I and II is
 cc_ex_lvl and if any indices correspond to RAS I or IV (i.e., include only all-active higher excitations)?
cc_ex_lvl and if any indices correspond to RAS I or IV (i.e., include only all-active higher excitations)?- Type: boolean
- Default: true
- CC_NUM_THREADS (CCENERGY)
CCENERGY — Number of threads
- Type: integer
- Default: 1
- CC_NUM_THREADS (CCEOM)
CCEOM — Number of threads
- Type: integer
- Default: 1
- CC_NUM_THREADS (CCTRIPLES)
CCTRIPLES — Number of threads
- Type: integer
- Default: 1
- CC_NUM_THREADS (PSIMRCC)
PSIMRCC — Number of threads
- Type: integer
- Default: 1
- CC_OS_SCALE (CCENERGY)
CCENERGY — Coupled-cluster opposite-spin scaling value
- Type: double
- Default: 1.27
- CC_RAS34_MAX (DETCI)
DETCI — maximum number of electrons in RAS III + IV, for CC
- Type: integer
- Default: -1
- CC_RAS3_MAX (DETCI)
DETCI — maximum number of electrons in RAS III, for CC
- Type: integer
- Default: -1
- CC_RAS4_MAX (DETCI)
DETCI — maximum number of electrons in RAS IV, for CC
- Type: integer
- Default: -1
- CC_SCALE_OS (FNOCC)
FNOCC — Oppposite-spin scaling factor for SCS-CCSD
- Type: double
- Default: 1.27
- CC_SCALE_SS (FNOCC)
FNOCC — Same-spin scaling factor for SCS-CCSD
- Type: double
- Default: 1.13
- CC_SS_SCALE (CCENERGY)
CCENERGY — Coupled-cluster same-spin scaling value
- Type: double
- Default: 1.13
- CC_TIMINGS (FNOCC)
FNOCC — Do time each cc diagram?
- Type: boolean
- Default: false
- CC_UPDATE_EPS (DETCI)
DETCI (Expert) — Do update T amplitudes with orbital eigenvalues? (Usually would do this). Not doing this is experimental.
- Type: boolean
- Default: true
- CC_VAL_EX_LEVEL (DETCI)
DETCI — The CC valence excitation level
- Type: integer
- Default: 0
- CC_VARIATIONAL (DETCI)
DETCI (Expert) — Do use variational energy expression in CC computation? Experimental.
- Type: boolean
- Default: false
- CC_VECS_READ (DETCI)
DETCI — Do import a CC vector from disk?
- Type: boolean
- Default: false
- CC_VECS_WRITE (DETCI)
DETCI — Do export a CC vector to disk?
- Type: boolean
- Default: false
- CCD_E_CONVERGENCE (SAPT)
SAPT — E converge value for CCD
- Type: conv double
- Default: 1e-8
- CCD_MAXITER (SAPT)
SAPT — Max CCD iterations
- Type: integer
- Default: 50
- CCD_T_CONVERGENCE (SAPT)
SAPT — Convergence tolerance for CCD amplitudes
- Type: conv double
- Default: 1e-8
- CCL_ENERGY (OCC)
OCC — Do compute CC Lambda energy? In order to this option to be valid one should use “TPDM_ABCD_TYPE = COMPUTE” option.
- Type: boolean
- Default: false
- CEPA_LEVEL (FNOCC)
FNOCC (Expert) — Which coupled-pair method is called? This parameter is used internally by the python driver. Changing its value won’t have any effect on the procedure.
- Type: string
- Default: CEPA(0)
- CEPA_NO_SINGLES (FNOCC)
FNOCC — Flag to exclude singly excited configurations from a coupled-pair computation.
- Type: boolean
- Default: false
- CEPA_OS_SCALE (OCC)
OCC — CEPA opposite-spin scaling value from SCS-CCSD
- Type: double
- Default: 1.27
- CEPA_SOS_SCALE (OCC)
OCC — CEPA Spin-opposite scaling (SOS) value
- Type: double
- Default: 1.3
- CEPA_SS_SCALE (OCC)
OCC — CEPA same-spin scaling value from SCS-CCSD
- Type: double
- Default: 1.13
- CEPA_TYPE (OCC)
OCC — CEPA type such as CEPA0, CEPA1 etc. currently we have only CEPA0.
- Type: string
- Possible Values: CEPA0
- Default: CEPA0
- CHECK_C_ORTHONORM (TRANSQT)
TRANSQT — Do check MO orthogonality condition?
- Type: boolean
- Default: false
- CHOLESKY_TOLERANCE (FNOCC)
FNOCC — tolerance for Cholesky decomposition of the ERI tensor
- Type: conv double
- Default: 1.0e-4
- CI_DIIS (MCSCF)
MCSCF — Do use DIIS extrapolation to accelerate convergence of the CI coefficients?
- Type: boolean
- Default: false
- CI_NUM_THREADS (DETCI)
DETCI — Number of threads for DETCI.
- Type: integer
- Default: 1
- CIBLKS_PRINT (DETCI)
DETCI — Do print a summary of the CI blocks?
- Type: boolean
- Default: false
- CIS_AD_STATES (CPHF)
CPHF — Which states to save AD Matrices for? * Positive - Singlets * Negative - Triplets *
- Type: array
- Default: No Default
- CIS_AMPLITUDE_CUTOFF (CPHF)
CPHF — Minimum singles amplitude to print in CIS analysis
- Type: double
- Default: 0.15
- CIS_DOPDM_STATES (CPHF)
CPHF — Which states to save AO difference OPDMs for? * Positive - Singlets * Negative - Triplets *
- Type: array
- Default: No Default
- CIS_MEM_SAFETY_FACTOR (CPHF)
CPHF — Memory safety factor for allocating JK
- Type: double
- Default: 0.75
- CIS_NO_STATES (CPHF)
CPHF — Which states to save AO Natural Orbitals for? * Positive - Singlets * Negative - Triplets *
- Type: array
- Default: No Default
- CIS_OPDM_STATES (CPHF)
CPHF — Which states to save AO OPDMs for? * Positive - Singlets * Negative - Triplets *
- Type: array
- Default: No Default
- CIS_TOPDM_STATES (CPHF)
CPHF — Which states to save AO transition OPDMs for? * Positive - Singlets * Negative - Triplets *
- Type: array
- Default: No Default
- COLLAPSE_SIZE (DETCI)
DETCI — Gives the number of vectors to retain when the Davidson subspace is collapsed (see MAX_NUM_VECS). If greater than one, the collapsed subspace retains the best estimate of the CI vector for the previous n iterations. Defaults to 1.
- Type: integer
- Default: 1
- COLLAPSE_WITH_LAST (CCEOM)
CCEOM — Do collapse with last vector?
- Type: boolean
- Default: true
- COMPLEX_TOLERANCE (CCEOM)
CCEOM — Complex tolerance applied in CCEOM computations
- Type: conv double
- Default: 1e-12
- COMPUTE_MP4_TRIPLES (FNOCC)
FNOCC (Expert) — Do compute MP4 triples contribution?
- Type: boolean
- Default: false
- COMPUTE_TRIPLES (FNOCC)
FNOCC (Expert) — Do compute triples contribution?
- Type: boolean
- Default: true
- CONSECUTIVE_BACKSTEPS (OPTKING)
OPTKING — Set number of consecutive backward steps allowed in optimization
- Type: integer
- Default: 0
- CORR_ANSATZ (PSIMRCC)
PSIMRCC — The ansatz to use for MRCC computations
- Type: string
- Possible Values: SR, MK, BW, APBW
- Default: MK
- CORR_CCSD_T (PSIMRCC)
PSIMRCC — The type of CCSD(T) computation to perform
- Type: string
- Possible Values: STANDARD, PITTNER
- Default: STANDARD
- CORR_CHARGE (PSIMRCC)
PSIMRCC — The molecular charge of the target state
- Type: integer
- Default: 0
- CORR_MULTP (PSIMRCC)
PSIMRCC — The multiplicity,
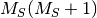 , of the target state. Must be specified if different from the reference
, of the target state. Must be specified if different from the reference 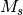 .
.- Type: integer
- Default: 1
- CORR_WFN (PSIMRCC)
PSIMRCC — The type of correlated wavefunction
- Type: string
- Possible Values: PT2, CCSD, MP2-CCSD, CCSD_T
- Default: CCSD
- COUPLING (PSIMRCC)
PSIMRCC — The order of coupling terms to include in MRCCSDT computations
- Type: string
- Possible Values: NONE, LINEAR, QUADRATIC, CUBIC
- Default: CUBIC
- COUPLING_TERMS (PSIMRCC)
PSIMRCC — Do include the terms that couple the reference determinants?
- Type: boolean
- Default: true
- COVALENT_CONNECT (OPTKING)
OPTKING — When determining connectivity, a bond is assigned if interatomic distance is less than (this number) * sum of covalent radii.
- Type: double
- Default: 1.3
- CPHF_MEM_SAFETY_FACTOR (CPHF)
CPHF — Memory safety factor for allocating JK
- Type: double
- Default: 0.75
- CPHF_TASKS (CPHF)
CPHF — Which tasks to run CPHF For * Valid choices: * -Polarizability *
- Type: array
- Default: No Default
- CUTOFF (OCC)
OCC — Cutoff value for numerical procedures
- Type: integer
- Default: 14
- D_CONVERGENCE (DFTSAPT)
DFTSAPT — Convergence criterion for residual of the CPKS coefficients in the SAPT *
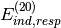 term.
term.- Type: conv double
- Default: 1e-8
- D_CONVERGENCE (MCSCF)
MCSCF — Convergence criterion for density.
- Type: conv double
- Default: 1e-6
- D_CONVERGENCE (SAPT)
SAPT — Convergence criterion for residual of the CPHF coefficients in the SAPT
term.- Type: conv double
- Default: 1e-8
- D_CONVERGENCE (SCF)
SCF — Convergence criterion for SCF density, which is defined as the RMS value of the orbital gradient. See Table SCF Convergence & Algorithm for default convergence criteria for different calculation types.
- Type: conv double
- Default: 1e-8
- DAMPING_CONVERGENCE (SCF)
SCF — The density convergence threshold after which damping is no longer performed, if it is enabled. It is recommended to leave damping on until convergence, which is the default.
- Type: conv double
- Default: 1.0e-18
- DAMPING_PERCENTAGE (DCFT)
DCFT (Expert) — The amount (percentage) of damping to apply to the orbital update procedure: 0 will result in a full update, 100 will completely stall the update. A value around 20 (which corresponds to 20% of the previous iteration’s density being mixed into the current iteration) can help in cases where oscillatory convergence is observed.
- Type: double
- Default: 0.0
- DAMPING_PERCENTAGE (PSIMRCC)
PSIMRCC — The amount (percentage) of damping to apply to the amplitude updates. 0 will result in a full update, 100 will completely stall the update. A value around 20 (which corresponds to 20% of the amplitudes from the previous iteration being mixed into the current iteration) can help in cases where oscillatory convergence is observed.
- Type: double
- Default: 0.0
- DAMPING_PERCENTAGE (SCF)
SCF — The amount (percentage) of damping to apply to the early density updates. 0 will result in a full update, 100 will completely stall the update. A value around 20 (which corresponds to 20% of the previous iteration’s density being mixed into the current density) could help to solve problems with oscillatory convergence.
- Type: double
- Default: 100.0
- DCFT_FUNCTIONAL (DCFT)
DCFT — Chooses appropriate DCFT method
- Type: string
- Possible Values: DC-06, DC-12, CEPA0
- Default: DC-06
- DCFT_GUESS (DCFT)
DCFT (Expert) — Whether to read the orbitals from a previous computation, or to compute an MP2 guess
- Type: string
- Possible Values: CC, BCC, MP2
- Default: MP2
- DEBUG (CPHF)
CPHF — The amount of debug information printed to the output file
- Type: integer
- Default: 0
- DEBUG (DFTSAPT)
DFTSAPT — Debug level
- Type: integer
- Default: 0
- DEBUG (GLOBALS)
GLOBALS (Expert) — The amount of information to print to the output file
- Type: integer
- Default: 0
- DELETE_AO (TRANSQT)
TRANSQT — Do delete AO integral files?
- Type: boolean
- Default: true
- DELETE_RESTR_DOCC (TRANSQT)
TRANSQT — Do delete restricted doubly occupieds?
- Type: boolean
- Default: true
- DELETE_TEI (TRANSQT2)
TRANSQT2 — Boolean to delete the SO-basis two-electron integral file after the transformation
- Type: boolean
- Default: true
- DELETE_TPDM (TRANSQT)
TRANSQT — Do delete TPDM file?
- Type: boolean
- Default: true
- DENOMINATOR_ALGORITHM (SAPT)
SAPT — Denominator algorithm for PT methods. Laplace transformations are slightly more efficient.
- Type: string
- Possible Values: LAPLACE, CHOLESKY
- Default: LAPLACE
- DENOMINATOR_DELTA (SAPT)
SAPT — Maximum error allowed (Max error norm in Delta tensor) in the approximate energy denominators employed for most of the
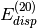 and
and 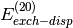 evaluation.
evaluation.- Type: double
- Default: 1.0e-6
- DERTYPE (GLOBALS)
GLOBALS (Expert) — Derivative level
- Type: string
- Possible Values: NONE, FIRST, SECOND, RESPONSE
- Default: NONE
- DETCI_FREEZE_CORE (DETCI)
DETCI — Do freeze core orbitals?
- Type: boolean
- Default: true
- DF_BASIS_CC (FNOCC)
FNOCC — Auxilliary basis for df-ccsd(t).
- Type: string
- Possible Values: basis string
- Default: No Default
- DF_BASIS_ELST (DFTSAPT)
DFTSAPT — The name of the electrostatic/exchange auxiliary basis set
- Type: string
- Possible Values: basis string
- Default: No Default
- DF_BASIS_ELST (SAPT)
SAPT — Auxiliary basis set for SAPT Elst10 and Exch10 density fitting computations, may be important if heavier elements are involved. Defaults to DF_BASIS_SAPT.
- Type: string
- Possible Values: basis string
- Default: No Default
- DF_BASIS_GUESS (SCF)
SCF — When BASIS_GUESS is active, run the preliminary scf in density-fitted mode with this as fitting basis for the small basis set. A value of TRUE turns on density fitting with the cc-pVDZ-RI basis set (when available for all elements).
- Type: string
- Possible Values: basis string
- Default: FALSE
- DF_BASIS_MP2 (DFMP2)
DFMP2 — Auxiliary basis set for MP2 density fitting computations. Defaults to a RI basis.
- Type: string
- Possible Values: basis string
- Default: No Default
- DF_BASIS_MP2 (LMP2)
LMP2 — Auxiliary basis set for MP2 density fitting calculations
- Type: string
- Possible Values: basis string
- Default: No Default
- DF_BASIS_SAPT (DFTSAPT)
DFTSAPT — The name of the response auxiliary basis set
- Type: string
- Possible Values: basis string
- Default: No Default
- DF_BASIS_SAPT (SAPT)
SAPT — Auxiliary basis set for SAPT density fitting computations. Defaults to a RI basis.
- Type: string
- Possible Values: basis string
- Default: No Default
- DF_BASIS_SCF (CPHF)
CPHF — Auxiliary basis for SCF
- Type: string
- Possible Values: basis string
- Default: No Default
- DF_BASIS_SCF (SCF)
SCF — Auxiliary basis set for SCF density fitting computations. Defaults to a JKFIT basis.
- Type: string
- Possible Values: basis string
- Default: No Default
- DF_FITTING_CONDITION (SCF)
SCF (Expert) — Fitting Condition
- Type: double
- Default: 1.0e-12
- DF_INTS_IO (DFMP2)
DFMP2 (Expert) — IO caching for CP corrections, etc
- Type: string
- Possible Values: NONE, SAVE, LOAD
- Default: NONE
- DF_INTS_IO (SCF)
SCF (Expert) — IO caching for CP corrections, etc
- Type: string
- Possible Values: NONE, SAVE, LOAD
- Default: NONE
- DF_INTS_NUM_THREADS (DFMP2)
DFMP2 — Number of threads to compute integrals with. 0 is wild card
- Type: integer
- Default: 0
- DF_INTS_NUM_THREADS (SCF)
SCF — Number of threads for integrals (may be turned down if memory is an issue). 0 is blank
- Type: integer
- Default: 0
- DF_LMP2 (LMP2)
LMP2 — Do use density fitting? Turned on with specification of fitting basis.
- Type: boolean
- Default: true
- DF_SCF_GUESS (SCF)
SCF — Use DF integrals tech to converge the SCF before switching to a conventional tech
- Type: boolean
- Default: true
- DFCC (FNOCC)
FNOCC — Do use density fitting in CC? This keyword is used internally by the driver. Changing its value will have no effect on the computation.
- Type: boolean
- Default: false
- DFMP2_MEM_FACTOR (DFMP2)
DFMP2 — % of memory for DF-MP2 three-index buffers
- Type: double
- Default: 0.9
- DFMP2_P2_TOLERANCE (DFMP2)
DFMP2 — Minimum error in the 2-norm of the P(2) matrix for corrections to Lia and P.
- Type: conv double
- Default: 0.0
- DFMP2_P_TOLERANCE (DFMP2)
DFMP2 — Minimum error in the 2-norm of the P matrix for skeleton-core Fock matrix derivatives.
- Type: conv double
- Default: 0.0
- DFT_ALPHA (SCF)
SCF — The DFT Exact-exchange parameter
- Type: double
- Default: 0.0
- DFT_BASIS_TOLERANCE (SCF)
SCF — DFT basis cutoff.
- Type: conv double
- Default: 1.0e-12
- DFT_BLOCK_MAX_POINTS (SCF)
SCF (Expert) — The maximum number of grid points per evaluation block.
- Type: integer
- Default: 5000
- DFT_BLOCK_MAX_RADIUS (SCF)
SCF (Expert) — The maximum radius to terminate subdivision of an octree block [au].
- Type: double
- Default: 3.0
- DFT_BLOCK_MIN_POINTS (SCF)
SCF (Expert) — The minimum number of grid points per evaluation block.
- Type: integer
- Default: 1000
- DFT_BLOCK_SCHEME (SCF)
SCF (Expert) — The blocking scheme for DFT.
- Type: string
- Possible Values: NAIVE, OCTREE
- Default: OCTREE
- DFT_BS_RADIUS_ALPHA (SCF)
SCF — Factor for effective BS radius in radial grid.
- Type: double
- Default: 1.0
- DFT_CUSTOM_FUNCTIONAL (SCF)
SCF — A custom DFT functional object (built by Python or NULL/None)
- Type: python
- Default: No Default
- DFT_DISPERSION_PARAMETERS (SCF)
SCF — Parameters defining the dispersion correction. See Table -D Functionals for default values and Table Dispersion Corrections for the order in which parameters are to be specified in this array option.
- Type: array
- Default: No Default
- DFT_FUNCTIONAL (SCF)
SCF — The DFT combined functional name, e.g. B3LYP, or GEN to use a python reference to a custom functional specified by DFT_CUSTOM_FUNCTIONAL.
- Type: string
- Default: No Default
- DFT_GRID_NAME (SCF)
SCF (Expert) — The DFT grid specification, such as SG1.
- Type: string
- Possible Values: SG1
- Default: No Default
- DFT_NUCLEAR_SCHEME (SCF)
SCF — Nuclear Scheme.
- Type: string
- Possible Values: TREUTLER, BECKE, NAIVE, STRATMANN
- Default: TREUTLER
- DFT_OMEGA (SCF)
SCF — The DFT Range-separation parameter
- Type: double
- Default: 0.0
- DFT_PRUNING_ALPHA (SCF)
SCF (Expert) — Spread alpha for logarithmic pruning.
- Type: double
- Default: 1.0
- DFT_PRUNING_SCHEME (SCF)
SCF (Expert) — Pruning Scheme.
- Type: string
- Possible Values: FLAT, P_GAUSSIAN, D_GAUSSIAN, P_SLATER, D_SLATER, LOG_GAUSSIAN, LOG_SLATER
- Default: FLAT
- DFT_RADIAL_POINTS (SCF)
SCF — Number of radial points.
- Type: integer
- Default: 75
- DFT_RADIAL_SCHEME (SCF)
SCF — Radial Scheme.
- Type: string
- Possible Values: TREUTLER, BECKE, MULTIEXP, EM, MURA
- Default: TREUTLER
- DFT_SPHERICAL_POINTS (SCF)
SCF — Number of spherical points (A Lebedev Points number).
- Type: integer
- Default: 302
- DFT_SPHERICAL_SCHEME (SCF)
SCF — Spherical Scheme.
- Type: string
- Possible Values: LEBEDEV
- Default: LEBEDEV
- DIAG_METHOD (CIS)
CIS — Diagonalization method for the CI matrix
- Type: string
- Possible Values: DAVIDSON, FULL
- Default: DAVIDSON
- DIAG_METHOD (DETCI)
DETCI — This specifies which method is to be used in diagonalizing the Hamiltonian. The valid options are: RSP, to form the entire H matrix and diagonalize using libciomr to obtain all eigenvalues (n.b. requires HUGE memory); OLSEN, to use Olsen’s preconditioned inverse subspace method (1990); MITRUSHENKOV, to use a 2x2 Olsen/Davidson method; and DAVIDSON (or SEM) to use Liu’s Simultaneous Expansion Method, which is identical to the Davidson method if only one root is to be found. There also exists a SEM debugging mode, SEMTEST. The SEM method is the most robust, but it also requires
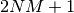 CI vectors on disk, where
CI vectors on disk, where 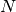 is the maximum number of iterations and
is the maximum number of iterations and 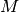 is the number of roots.
is the number of roots.- Type: string
- Possible Values: RSP, OLSEN, MITRUSHENKOV, DAVIDSON, SEM, SEMTEST
- Default: SEM
- DIAGONAL_CCSD_T (PSIMRCC)
PSIMRCC — Do include the diagonal corrections in (T) computations?
- Type: boolean
- Default: true
- DIAGONALIZE_HEFF (PSIMRCC)
PSIMRCC — Do diagonalize the effective Hamiltonian?
- Type: boolean
- Default: false
- DIE_IF_NOT_CONVERGED (GLOBALS)
GLOBALS (Expert) — PSI4 dies if energy does not converge.
- Type: boolean
- Default: true
- DIIS (CCENERGY)
CCENERGY — Do use DIIS extrapolation to accelerate convergence?
- Type: boolean
- Default: true
- DIIS (CCLAMBDA)
CCLAMBDA — Do use DIIS extrapolation to accelerate convergence?
- Type: boolean
- Default: true
- DIIS (CCRESPONSE)
CCRESPONSE — Do use DIIS extrapolation to accelerate convergence?
- Type: boolean
- Default: true
- DIIS (DETCI)
DETCI — Do use DIIS extrapolation to accelerate CC convergence?
- Type: boolean
- Default: true
- DIIS (LMP2)
LMP2 — Do use DIIS extrapolation to accelerate convergence?
- Type: boolean
- Default: true
- DIIS (MCSCF)
MCSCF — Do use DIIS extrapolation to accelerate convergence of the SCF energy (MO coefficients only)?
- Type: boolean
- Default: true
- DIIS (SCF)
SCF — Do use DIIS extrapolation to accelerate convergence?
- Type: boolean
- Default: true
- DIIS_FREQ (DETCI)
DETCI — How often to do a DIIS extrapolation. 1 means do DIIS every iteration, 2 is every other iteration, etc.
- Type: integer
- Default: 1
- DIIS_MAX_VECS (DCFT)
DCFT (Expert) — Maximum number of error vectors stored for DIIS extrapolation
- Type: integer
- Default: 6
- DIIS_MAX_VECS (DETCI)
DETCI — Maximum number of error vectors stored for DIIS extrapolation
- Type: integer
- Default: 5
- DIIS_MAX_VECS (FNOCC)
FNOCC — Desired number of DIIS vectors
- Type: integer
- Default: 8
- DIIS_MAX_VECS (LMP2)
LMP2 — Maximum number of error vectors stored for DIIS extrapolation
- Type: integer
- Default: 5
- DIIS_MAX_VECS (MCSCF)
MCSCF — Maximum number of error vectors stored for DIIS extrapolation
- Type: integer
- Default: 7
- DIIS_MAX_VECS (PSIMRCC)
PSIMRCC — Maximum number of error vectors stored for DIIS extrapolation
- Type: integer
- Default: 7
- DIIS_MAX_VECS (SCF)
SCF — Maximum number of error vectors stored for DIIS extrapolation
- Type: integer
- Default: 10
- DIIS_MIN_VECS (DCFT)
DCFT (Expert) — Minimum number of error vectors stored for DIIS extrapolation
- Type: integer
- Default: 3
- DIIS_MIN_VECS (DETCI)
DETCI — Minimum number of error vectors stored for DIIS extrapolation
- Type: integer
- Default: 2
- DIIS_MIN_VECS (SCF)
SCF — Minimum number of error vectors stored for DIIS extrapolation
- Type: integer
- Default: 2
- DIIS_START (PSIMRCC)
PSIMRCC — The number of DIIS vectors needed before extrapolation is performed
- Type: integer
- Default: 2
- DIIS_START (SCF)
SCF — The minimum iteration to start storing DIIS vectors
- Type: integer
- Default: 1
- DIIS_START_CONVERGENCE (DCFT)
DCFT — Value of RMS of the density cumulant residual and SCF error vector below which DIIS extrapolation starts. Same keyword controls the DIIS extrapolation for the solution of the response equations.
- Type: conv double
- Default: 1e-3
- DIIS_START_ITER (DETCI)
DETCI — Iteration at which to start using DIIS
- Type: integer
- Default: 1
- DIIS_START_ITER (LMP2)
LMP2 — Iteration at which to start DIIS extrapolation
- Type: integer
- Default: 3
- DIPMOM (DETCI)
DETCI — Do compute the dipole moment?
- Type: boolean
- Default: false
- DIPMOM (FNOCC)
FNOCC — Compute the dipole moment? Note that dipole moments are only available in the FNOCC module for the ACPF, AQCC, CISD, and CEPA(0) methods.
- Type: boolean
- Default: false
- DISP_SIZE (FINDIF)
FINDIF — Displacement size in au for finite-differences.
- Type: double
- Default: 0.005
- DISTANT_PAIR_CUTOFF (LMP2)
LMP2 — Distant pair cutoff
- Type: double
- Default: 8.0
- DISTRIBUTED_MATRIX (SCF)
SCF (Expert) — The dimension sizes of the distributed matrix
- Type: array
- Default: No Default
- DO_ALL_TEI (TRANSQT)
TRANSQT — Do transform all TEIs
- Type: boolean
- Default: false
- DO_CCD_DISP (SAPT)
SAPT (Expert) — Do CCD dispersion correction in SAPT2+, SAPT2+(3) or SAPT2+3?
- Type: boolean
- Default: false
- DO_DIIS (OCC)
OCC — Do apply DIIS extrapolation?
- Type: boolean
- Default: true
- DO_LEVEL_SHIFT (OCC)
OCC — Do apply level shifting?
- Type: boolean
- Default: true
- DO_SCS (OCC)
OCC — Do perform spin-component-scaled OMP2 (SCS-OMP2)? In all computation, SCS-OMP2 energy is computed automatically. However, in order to perform geometry optimizations and frequency computations with SCS-OMP2, one needs to set ‘DO_SCS’ to true
- Type: boolean
- Default: false
- DO_SINGLETS (CPHF)
CPHF — Do singlet states? Default true
- Type: boolean
- Default: true
- DO_SOS (OCC)
OCC — Do perform spin-opposite-scaled OMP2 (SOS-OMP2)? In all computation, SOS-OMP2 energy is computed automatically. However, in order to perform geometry optimizations and frequency computations with SOS-OMP2, one needs to set ‘DO_SOS’ to true
- Type: boolean
- Default: false
- DO_THIRD_ORDER (SAPT)
SAPT (Expert) — Do compute third-order corrections?
- Type: boolean
- Default: false
- DO_TRIPLETS (CPHF)
CPHF — Do triplet states? Default true
- Type: boolean
- Default: true
- DOCC (GLOBALS)
GLOBALS — An array containing the number of doubly-occupied orbitals per irrep (in Cotton order)
- Type: array
- Default: No Default
- DOCC (MCSCF)
MCSCF — The number of doubly occupied orbitals, per irrep
- Type: array
- Default: No Default
- DOMAIN_PRINT (CIS)
CIS — Do print the domains?
- Type: boolean
- Default: false
- DOMAIN_PRINT_EXIT (LMP2)
LMP2 — Do exit after printing the domains?
- Type: boolean
- Default: false
- DOMAINS (CIS)
CIS —
- Type: array
- Default: No Default
- E3_SCALE (OCC)
OCC — Scaling value for 3rd order energy correction (S. Grimme, Vol. 24, pp. 1529, J. Comput. Chem.)
- Type: double
- Default: 0.25
- E_CONVERGENCE (CCENERGY)
CCENERGY — Convergence criterion for energy. See Table Post-SCF Convergence for default convergence criteria for different calculation types.
- Type: conv double
- Default: 1e-8
- E_CONVERGENCE (CCEOM)
CCEOM — Convergence criterion for excitation energy (change) in the Davidson algorithm for CC-EOM. See Table Post-SCF Convergence for default convergence criteria for different calculation types.
- Type: conv double
- Default: 1e-8
- E_CONVERGENCE (DETCI)
DETCI — Convergence criterion for energy. See Table Post-SCF Convergence for default convergence criteria for different calculation types.
- Type: conv double
- Default: 1e-8
- E_CONVERGENCE (FNOCC)
FNOCC — Convergence criterion for CC energy. See Table Post-SCF Convergence for default convergence criteria for different calculation types. Note that convergence is met only when E_CONVERGENCE and R_CONVERGENCE are satisfied.
- Type: conv double
- Default: 1.0e-8
- E_CONVERGENCE (LMP2)
LMP2 — Convergence criterion for energy (change). See Table Post-SCF Convergence for default convergence criteria for different calculation types.
- Type: conv double
- Default: 1e-8
- E_CONVERGENCE (MCSCF)
MCSCF — Convergence criterion for energy.
- Type: conv double
- Default: 1e-8
- E_CONVERGENCE (MRCC)
MRCC — Convergence criterion for energy. See Table Post-SCF Convergence for default convergence criteria for different calculation types. This becomes tol (option #16) in fort.56.
- Type: conv double
- Default: 1e-8
- E_CONVERGENCE (OCC)
OCC — Convergence criterion for energy. See Table Post-SCF Convergence for default convergence criteria for different calculation types.
- Type: conv double
- Default: 1e-8
- E_CONVERGENCE (PSIMRCC)
PSIMRCC — Convergence criterion for energy. See Table Post-SCF Convergence for default convergence criteria for different calculation types.
- Type: conv double
- Default: 1e-8
- E_CONVERGENCE (SAPT)
SAPT — Convergence criterion for energy (change) in the SAPT
term during solution of the CPHF equations.- Type: conv double
- Default: 1e-10
- E_CONVERGENCE (SCF)
SCF — Convergence criterion for SCF energy. See Table SCF Convergence & Algorithm for default convergence criteria for different calculation types.
- Type: conv double
- Default: 1e-8
- EA_POLES (OCC)
OCC — Do compute OCC poles for electron affinities? Only valid for OMP2.
- Type: boolean
- Default: false
- EKT_EA (OCC)
OCC — Do compute virtual orbital energies based on extended Koopmans’ theorem?
- Type: boolean
- Default: false
- EKT_IP (OCC)
OCC — Do compute occupied orbital energies based on extended Koopmans’ theorem?
- Type: boolean
- Default: false
- EOM_GUESS (CCEOM)
CCEOM — Specifies a set of single-excitation guess vectors for the EOM-CC procedure. If EOM_GUESS = SINGLES, the guess will be taken from the singles-singles block of the similarity-transformed Hamiltonian, Hbar. If EOM_GUESS = DISK, guess vectors from a previous computation will be read from disk. If EOM_GUESS = INPUT, guess vectors will be specified in user input. The latter method is not currently available.
- Type: string
- Possible Values: SINGLES, DISK, INPUT
- Default: SINGLES
- EOM_REFERENCE (CCEOM)
CCEOM — Reference wavefunction type for EOM computations
- Type: string
- Possible Values: RHF, ROHF, UHF
- Default: RHF
- EOM_REFERENCE (CCHBAR)
CCHBAR — Reference wavefunction type for EOM computations
- Type: string
- Default: RHF
- EOM_REFERENCE (CCSORT)
CCSORT — Reference wavefunction type for EOM computations
- Type: string
- Default: RHF
- EP_EA_POLES (OCC)
OCC — Do compute EP-OCC poles for electron affinities? Only valid for OMP2.
- Type: boolean
- Default: false
- EP_IP_POLES (OCC)
OCC — Do compute EP-OCC poles for ionization potentials? Only valid OMP2.
- Type: boolean
- Default: false
- EP_MAXITER (OCC)
OCC — Maximum number of electron propagator iterations.
- Type: integer
- Default: 30
- EX_ALLOW (DETCI)
DETCI (Expert) — An array of length EX_LEVEL specifying whether each excitation type (S,D,T, etc.) is allowed (1 is allowed, 0 is disallowed). Used to specify non-standard CI spaces such as CIST.
- Type: array
- Default: No Default
- EX_LEVEL (DETCI)
DETCI — The CI excitation level
- Type: integer
- Default: 2
- EXCITATION_RANGE (CCEOM)
CCEOM (Expert) — The depth into the occupied and valence spaces from which one-electron excitations are seeded into the Davidson guess to the CIS (the default of 2 includes all single excitations between HOMO-1, HOMO, LUMO, and LUMO+1). This CIS is in turn the Davidson guess to the EOM-CC. Expand to capture more exotic excited states in the EOM-CC calculation
- Type: integer
- Default: 2
- EXPLICIT_HAMILTONIAN (CPHF)
CPHF — Do explicit hamiltonian only?
- Type: boolean
- Default: false
- EXTERN (SCF)
SCF — An ExternalPotential (built by Python or NULL/None)
- Type: python
- Default: No Default
- FAIL_ON_MAXITER (SCF)
SCF — Fail if we reach maxiter without converging?
- Type: boolean
- Default: true
- FAVG (MCSCF)
MCSCF — Do use the average Fock matrix during the SCF optimization?
- Type: boolean
- Default: false
- FAVG_CCSD_T (PSIMRCC)
PSIMRCC — Do use the averaged Fock matrix over all references in (T) computations?
- Type: boolean
- Default: false
- FAVG_START (MCSCF)
MCSCF — Iteration at which to begin using the averaged Fock matrix
- Type: integer
- Default: 5
- FCI (DETCI)
DETCI — Do a full CI (FCI)? If TRUE, overrides the value of EX_LEVEL.
- Type: boolean
- Default: false
- FCI_STRINGS (DETCI)
DETCI (Expert) — Do store strings specifically for FCI? (Defaults to TRUE for FCI.)
- Type: boolean
- Default: false
- FILTER_GUESS (DETCI)
DETCI (Expert) — Do invoke the FILTER_GUESS options that are used to filter out some trial vectors which may not have the appropriate phase convention between two determinants? This is useful to remove, e.g., delta states when a sigma state is desired. The user inputs two determinants (by giving the absolute alpha string number and beta string number for each), and also the desired phase between these two determinants for guesses which are to be kept. FILTER_GUESS = TRUE turns on the filtering routine. Requires additional keywords FILTER_GUESS_DET1, FILTER_GUESS_DET2, and FILTER_GUESS_SIGN.
- Type: boolean
- Default: false
- FILTER_GUESS_DET1 (DETCI)
DETCI (Expert) — Array specifying the absolute alpha string number and beta string number for the first determinant in the filter procedure. (See FILTER_GUESS).
- Type: array
- Default: No Default
- FILTER_GUESS_DET2 (DETCI)
DETCI (Expert) — Array specifying the absolute alpha string number and beta string number for the second determinant in the filter procedure. (See FILTER_GUESS).
- Type: array
- Default: No Default
- FILTER_GUESS_SIGN (DETCI)
DETCI (Expert) — The required phase (1 or -1) between the two determinants specified by FILTER_GUESS_DET1 and FILTER_GUESS_DET2.
- Type: integer
- Default: 1
- FILTER_ZERO_DET (DETCI)
DETCI (Expert) — If present, the code will try to filter out a particular determinant by setting its CI coefficient to zero. FILTER_ZERO_DET = [alphastr, betastr] specifies the absolute alpha and beta string numbers of the target determinant. This could be useful for trying to exclude states that have a nonzero CI coefficient for the given determinant. However, this option was experimental and may not be effective.
- Type: array
- Default: No Default
- FINAL_GEOM_WRITE (OPTKING)
OPTKING — Do save and print the geometry from the last projected step at the end of a geometry optimization? Otherwise (and by default), save and print the previous geometry at which was computed the gradient that satisfied the convergence criteria.
- Type: boolean
- Default: false
- FIRST_TMP_FILE (TRANSQT)
TRANSQT — First temporary file
- Type: integer
- Default: 150
- FITTING_ALGORITHM (CPHF)
CPHF — Fitting algorithm (0 for old, 1 for new)
- Type: integer
- Default: 0
- FITTING_CONDITION (CPHF)
CPHF — The maximum reciprocal condition allowed in the fitting metric
- Type: double
- Default: 1.0e-12
- FLEXIBLE_G_CONVERGENCE (OPTKING)
OPTKING — Even if a user-defined threshold is set, allow for normal, flexible convergence criteria
- Type: boolean
- Default: false
- FOCK_TOLERANCE (LMP2)
LMP2 — Minimum absolute value below which parts of the Fock matrix are skipped.
- Type: conv double
- Default: 1e-2
- FOLLOW (STABILITY)
STABILITY — Do follow the most negative eigenvalue of the Hessian towards a lower energy HF solution? Follow a UHF
 UHF instability of same symmetry?
UHF instability of same symmetry?- Type: boolean
- Default: false
- FOLLOW_ROOT (CLAG)
CLAG — Root to get OPDM
- Type: integer
- Default: 1
- FOLLOW_ROOT (DETCI)
DETCI — The root to write out the two-particle density matrix for (the one-particle density matrices are written for all roots). Useful for a state-specific CASSCF or CI optimization on an excited state.
- Type: integer
- Default: 1
- FOLLOW_ROOT (MCSCF)
MCSCF — Which solution of the SCF equations to find, where 1 is the SCF ground state
- Type: integer
- Default: 1
- FOLLOW_ROOT (PSIMRCC)
PSIMRCC — Which root of the effective hamiltonian is the target state?
- Type: integer
- Default: 1
- FOLLOW_STEP_SCALE (SCF)
SCF (Expert) — When using STABILITY_ANALYSIS = FOLLOW, how much to scale the step along the eigenvector by.
- Type: double
- Default: 0.5
- FOLLOW_VECTOR (DETCI)
DETCI (Expert) — In following a particular root (see FOLLOW_ROOT), sometimes the root number changes. To follow a root of a particular character, one can specify a list of determinants and their coefficients, and the code will follow the root with the closest overlap. The user specifies arrays containing the absolute alpha string indices (A_i below), absolute beta indices (B_i below), and CI coefficients (C_i below) to form the desired vector. The format is FOLLOW_VECTOR = [ [[A_1, B_1], C_1], [[A_2, B_2], C_2], ...].
- Type: array
- Default: No Default
- FORCE_RESTART (CCENERGY)
CCENERGY (Expert) — Do restart the coupled-cluster iterations even if MO phases are screwed up?
- Type: boolean
- Default: false
- FORCE_TWOCON (MCSCF)
MCSCF — Do attempt to force a two configruation solution by starting with CI coefficents of
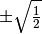 ?
?- Type: boolean
- Default: false
- FRAC_DIIS (SCF)
SCF — Do use DIIS extrapolation to accelerate convergence in frac?
- Type: boolean
- Default: true
- FRAC_LOAD (SCF)
SCF — Do recompute guess from stored orbitals?
- Type: boolean
- Default: false
- FRAC_OCC (SCF)
SCF — The absolute indices of occupied orbitals to fractionally occupy (+/- for alpha/beta)
- Type: array
- Default: No Default
- FRAC_RENORMALIZE (SCF)
SCF — Do renormalize C matrices prior to writing to checkpoint?
- Type: boolean
- Default: true
- FRAC_START (SCF)
SCF — The iteration to start fractionally occupying orbitals (or 0 for no fractional occupation)
- Type: integer
- Default: 0
- FRAC_VAL (SCF)
SCF — The occupations of the orbital indices specified above (
 )
)- Type: array
- Default: No Default
- FRAG_MODE (OPTKING)
OPTKING — For multi-fragment molecules, treat as single bonded molecule or via interfragment coordinates. A primary difference is that in MULTI mode, the interfragment coordinates are not redundant.
- Type: string
- Possible Values: SINGLE, MULTI
- Default: SINGLE
- FREEZE_CORE (GLOBALS)
GLOBALS — Specifies how many core orbitals to freeze in correlated computations. TRUE will default to freezing the standard default number of core orbitals. For PSI, the standard number of core orbitals is the number of orbitals in the nearest previous noble gas atom. More precise control over the number of frozen orbitals can be attained by using the keywords NUM_FROZEN_DOCC (gives the total number of orbitals to freeze, program picks the lowest-energy orbitals) or FROZEN_DOCC (gives the number of orbitals to freeze per irreducible representation)
- Type: string
- Possible Values: FALSE, TRUE
- Default: FALSE
- FREEZE_CORE (SAPT)
SAPT — The scope of core orbitals to freeze in evaluation of SAPT
and terms. Recommended true for all SAPT computations- Type: string
- Possible Values: FALSE, TRUE
- Default: FALSE
- FREEZE_INTERFRAG (OPTKING)
OPTKING — Do freeze all interfragment modes?
- Type: boolean
- Default: false
- FREEZE_INTRAFRAG (OPTKING)
OPTKING — Do freeze all fragments rigid?
- Type: boolean
- Default: false
- FROZEN_BEND (OPTKING)
OPTKING — Specify angles between atoms to be frozen
- Type: string
- Default: No Default
- FROZEN_DIHEDRAL (OPTKING)
OPTKING — Specify dihedral angles between atoms to be frozen
- Type: string
- Default: No Default
- FROZEN_DISTANCE (OPTKING)
OPTKING — Specify distances between atoms to be frozen
- Type: string
- Default: No Default
- FROZEN_DOCC (GLOBALS)
GLOBALS — An array containing the number of frozen doubly-occupied orbitals per irrep (these are not excited in a correlated wavefunction, nor can they be optimized in MCSCF. This trumps NUM_FROZEN_DOCC and FREEZE_CORE.
- Type: array
- Default: No Default
- FROZEN_DOCC (PSIMRCC)
PSIMRCC — The number of frozen occupied orbitals per irrep
- Type: array
- Default: No Default
- FROZEN_UOCC (GLOBALS)
GLOBALS — An array containing the number of frozen unoccupied orbitals per irrep (these are not populated in a correlated wavefunction, nor can they be optimized in MCSCF. This trumps NUM_FROZEN_UOCC.
- Type: array
- Default: No Default
- FROZEN_UOCC (PSIMRCC)
PSIMRCC — The number of frozen virtual orbitals per irrep
- Type: array
- Default: No Default
- FULL_HESS_EVERY (OPTKING)
OPTKING — Frequency with which to compute the full Hessian in the course of a geometry optimization. 0 means to compute the initial Hessian only, 1 means recompute every step, and N means recompute every N steps. The default (-1) is to never compute the full Hessian.
- Type: integer
- Default: -1
- FULL_MATRIX (CCEOM)
CCEOM — Do use full effective Hamiltonian matrix?
- Type: boolean
- Default: false
- FZC_A_FILE (TRANSQT)
TRANSQT — Alpha-spin frozen-core file
- Type: integer
- Default: PSIF_OEI
- FZC_B_FILE (TRANSQT)
TRANSQT — Beta-spin frozen-core file
- Type: integer
- Default: PSIF_OEI
- FZC_FILE (TRANSQT)
TRANSQT — Frozen-core file
- Type: integer
- Default: PSIF_OEI
- G_CONVERGENCE (OPTKING)
OPTKING — Set of optimization criteria. Specification of any MAX_*_G_CONVERGENCE or RMS_*_G_CONVERGENCE options will append to overwrite the criteria set here unless FLEXIBLE_G_CONVERGENCE is also on. See Table Geometry Convergence for details.
- Type: string
- Possible Values: QCHEM, MOLPRO, GAU, GAU_LOOSE, GAU_TIGHT, GAU_VERYTIGHT, TURBOMOLE, CFOUR, NWCHEM_LOOSE
- Default: QCHEM
- GAUGE (CCDENSITY)
CCDENSITY — The type of gauge to use for properties
- Type: string
- Default: LENGTH
- GAUGE (CCRESPONSE)
CCRESPONSE — Specifies the choice of representation of the electric dipole operator. Acceptable values are LENGTH for the usual length-gauge representation, VELOCITY for the modified velocity-gauge representation in which the static-limit optical rotation tensor is subtracted from the frequency- dependent tensor, or BOTH. Note that, for optical rotation calculations, only the choices of VELOCITY or BOTH will yield origin-independent results.
- Type: string
- Possible Values: LENGTH, VELOCITY, BOTH
- Default: LENGTH
- GEOM_MAXITER (OPTKING)
OPTKING — Maximum number of geometry optimization steps
- Type: integer
- Default: 50
- GRADIENT_WRITE (FINDIF)
FINDIF — Do write a gradient output file? If so, the filename will end in .grad, and the prefix is determined by WRITER_FILE_LABEL (if set), or else by the name of the output file plus the name of the current molecule.
- Type: boolean
- Default: false
- GUESS (SCF)
SCF — The type of guess orbitals. Defaults to CORE except for geometry optimizations, in which case READ becomes the default after the first geometry step.
- Type: string
- Possible Values: CORE, GWH, SAD, READ
- Default: CORE
- GUESS_VECTOR (DETCI)
DETCI (Expert) — Guess vector type. Accepted values are UNIT for a unit vector guess (NUM_ROOTS and NUM_INIT_VECS must both be 1); H0_BLOCK to use eigenvectors from the H0 BLOCK submatrix (default); DFILE to use NUM_ROOTS previously converged vectors in the D file; IMPORT to import a guess previously exported from a CI computation (possibly using a different CI space)
- Type: string
- Possible Values: UNIT, H0_BLOCK, DFILE, IMPORT
- Default: H0_BLOCK
- H0_BLOCK_COUPLING (DETCI)
DETCI (Expert) — Do use coupling block in preconditioner?
- Type: boolean
- Default: false
- H0_BLOCK_COUPLING_SIZE (DETCI)
DETCI (Expert) — Parameters which specifies the size of the coupling block within the generalized davidson preconditioner.
- Type: integer
- Default: 0
- H0_BLOCKSIZE (DETCI)
DETCI (Expert) — This parameter specifies the size of the H0 block of the Hamiltonian which is solved exactly. The n determinants with the lowest SCF energy are selected, and a submatrix of the Hamiltonian is formed using these determinants. This submatrix is used to accelerate convergence of the CI iterations in the OLSEN and MITRUSHENKOV iteration schemes, and also to find a good starting guess for the SEM method if GUESS_VECTOR is H0_BLOCK. Defaults to 400. Note that the program may change the given size for Ms=0 cases (MS0 is TRUE) if it determines that the H0 block includes only one member of a pair of determinants related by time reversal symmetry. For very small block sizes, this could conceivably eliminate the entire H0 block; the program should print warnings if this occurs.
- Type: integer
- Default: 400
- H0_GUESS_SIZE (DETCI)
DETCI (Expert) — size of H0 block for initial guess
- Type: integer
- Default: 400
- H_BOND_CONNECT (OPTKING)
OPTKING — For now, this is a general maximum distance for the definition of H-bonds
- Type: double
- Default: 4.3
- HD_AVG (DETCI)
DETCI (Expert) — How to average H diag energies over spin coupling sets. HD_EXACT uses the exact diagonal energies which results in expansion vectors which break spin symmetry. HD_KAVE averages the diagonal energies over a spin-coupling set yielding spin pure expansion vectors. ORB_ENER employs the sum of orbital energy approximation giving spin pure expansion vectors but usually doubles the number of Davidson iterations. EVANGELISTI uses the sums and differences of orbital energies with the SCF reference energy to produce spin pure expansion vectors. LEININGER approximation which subtracts the one-electron contribution from the orbital energies, multiplies by 0.5, and adds the one-electron contribution back in, producing spin pure expansion vectors and developed by Matt Leininger and works as well as EVANGELISTI.
- Type: string
- Possible Values: EVANGELISTI, HD_EXACT, HD_KAVE, ORB_ENER, LEININGER, Z_KAVE
- Default: EVANGELISTI
- HD_OTF (DETCI)
DETCI (Expert) — Do compute the diagonal elements of the Hamiltonian matrix on-the-fly? Otherwise, a diagonal element vector is written to a separate file on disk.
- Type: boolean
- Default: true
- HEFF4 (PSIMRCC)
PSIMRCC — Do include the fourth-order contributions to the effective Hamiltonian?
- Type: boolean
- Default: true
- HEFF_PRINT (PSIMRCC)
PSIMRCC — Do print the effective Hamiltonian?
- Type: boolean
- Default: false
- HESS_UPDATE (OPTKING)
OPTKING — Hessian update scheme
- Type: string
- Possible Values: NONE, BFGS, MS, POWELL, BOFILL
- Default: BFGS
- HESS_UPDATE_LIMIT (OPTKING)
OPTKING — Do limit the magnitude of changes caused by the Hessian update?
- Type: boolean
- Default: true
- HESS_UPDATE_LIMIT_MAX (OPTKING)
OPTKING — If HESS_UPDATE_LIMIT is true, changes to the Hessian from the update are limited to the larger of HESS_UPDATE_LIMIT_SCALE * (the previous value) and HESS_UPDATE_LIMIT_MAX [au].
- Type: double
- Default: 1.00
- HESS_UPDATE_LIMIT_SCALE (OPTKING)
OPTKING — If HESS_UPDATE_LIMIT is true, changes to the Hessian from the update are limited to the larger of HESS_UPDATE_LIMIT_SCALE * (the previous value) and HESS_UPDATE_LIMIT_MAX [au].
- Type: double
- Default: 0.50
- HESS_UPDATE_USE_LAST (OPTKING)
OPTKING — Number of previous steps to use in Hessian update, 0 uses all
- Type: integer
- Default: 1
- HESSIAN_WRITE (FINDIF)
FINDIF — Do write a hessian output file? If so, the filename will end in .hess, and the prefix is determined by WRITER_FILE_LABEL (if set), or else by the name of the output file plus the name of the current molecule.
- Type: boolean
- Default: false
- ICORE (DETCI)
DETCI — Specifies how to handle buffering of CI vectors. A value of 0 makes the program perform I/O one RAS subblock at a time; 1 uses entire CI vectors at a time; and 2 uses one irrep block at a time. Values of 0 or 2 cause some inefficiency in the I/O (requiring multiple reads of the C vector when constructing H in the iterative subspace if DIAG_METHOD = SEM), but require less core memory.
- Type: integer
- Default: 1
- IGNORE_TAU (DCFT)
DCFT (Expert) — Controls whether to ignore terms containing non-idempotent contribution to OPDM or not (for debug puproses only). For practical applications only the default must be used
- Type: boolean
- Default: false
- INTCO_FIXED_EQ_FORCE_CONSTANT (OPTKING)
OPTKING — In constrained optimizations, for internal coordinates with user-specified equilibrium values, this is the force constant (in au) used to apply an additional force to each coordinate. If the user is only concerned to satify the desired constraint, then the user need only ensure that this value is sufficiently large. Alternatively, the user may specify this value to apply a force of a particular magnitude, in which case the given equilibrium value may or may not be reached by the optimization.
- Type: double
- Default: 2.0
- INTCOS_GENERATE_EXIT (OPTKING)
OPTKING — Do only generate the internal coordinates and then stop?
- Type: boolean
- Default: false
- INTERFRAG_DIST_INV (OPTKING)
OPTKING — Do use
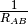 for the stretching coordinate between fragments? Otherwise, use
for the stretching coordinate between fragments? Otherwise, use 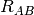 .
.- Type: boolean
- Default: false
- INTERFRAG_HESS (OPTKING)
OPTKING — Model Hessian to guess interfragment force constants
- Type: string
- Possible Values: DEFAULT, FISCHER_LIKE
- Default: DEFAULT
- INTERFRAG_MODE (OPTKING)
OPTKING — When interfragment coordinates are present, use as reference points either principal axes or fixed linear combinations of atoms.
- Type: string
- Possible Values: FIXED, INTERFRAGMENT
- Default: FIXED
- INTERFRAG_STEP_LIMIT (OPTKING)
OPTKING — Maximum step size in bohr or radian along an interfragment coordinate
- Type: double
- Default: 0.4
- INTERFRAGMENT_CONNECT (OPTKING)
OPTKING — When connecting disparate fragments when frag_mode = SIMPLE, a “bond” is assigned if interatomic distance is less than (this number) * sum of covalent radii. The value is then increased until all the fragments are connected (directly or indirectly).
- Type: double
- Default: 1.8
- INTERNAL_ROTATIONS (MCSCF)
MCSCF — Do consider internal rotations?
- Type: boolean
- Default: true
- INTRAFRAG_HESS (OPTKING)
OPTKING — Model Hessian to guess intrafragment force constants
- Type: string
- Possible Values: FISCHER, SCHLEGEL, SIMPLE, LINDH
- Default: SCHLEGEL
- INTRAFRAG_STEP_LIMIT (OPTKING)
OPTKING — Initial maximum step size in bohr or radian along an internal coordinate
- Type: double
- Default: 0.4
- INTRAFRAG_STEP_LIMIT_MAX (OPTKING)
OPTKING — Upper bound for dynamic trust radius [au]
- Type: double
- Default: 1.0
- INTRAFRAG_STEP_LIMIT_MIN (OPTKING)
OPTKING — Lower bound for dynamic trust radius [au]
- Type: double
- Default: 0.001
- INTS_TOLERANCE (CCDENSITY)
CCDENSITY — Minimum absolute value below which integrals are neglected.
- Type: conv double
- Default: 1e-14
- INTS_TOLERANCE (CCSORT)
CCSORT — Minimum absolute value below which integrals are neglected.
- Type: conv double
- Default: 1e-14
- INTS_TOLERANCE (DCFT)
DCFT (Expert) — Minimum absolute value below which integrals are neglected
- Type: conv double
- Default: 1e-14
- INTS_TOLERANCE (DFMP2)
DFMP2 — Minimum absolute value below which integrals are neglected.
- Type: conv double
- Default: 0.0
- INTS_TOLERANCE (LMP2)
LMP2 — Minimum absolute value below which integrals are neglected.
- Type: conv double
- Default: 1e-7
- INTS_TOLERANCE (MRCC)
MRCC — Minimum absolute value below which integrals are neglected.
- Type: conv double
- Default: 1.0e-12
- INTS_TOLERANCE (SAPT)
SAPT — Minimum absolute value below which all three-index DF integrals and those contributing to four-index integrals are neglected. The default is conservative, but there isn’t much to be gained from loosening it, especially for higher-order SAPT.
- Type: conv double
- Default: 1.0e-12
- INTS_TOLERANCE (SCF)
SCF — Minimum absolute value below which TEI are neglected.
- Type: conv double
- Default: 0.0
- INTS_TOLERANCE (TRANSQT)
TRANSQT — Minimum absolute value below which integrals are neglected.
- Type: conv double
- Default: 1e-14
- INTS_TOLERANCE (TRANSQT2)
TRANSQT2 — Minimum absolute value below which integrals are neglected.
- Type: conv double
- Default: 1e-14
- IP_POLES (OCC)
OCC — Do compute OCC poles for ionization potentials? Only valid OMP2.
- Type: boolean
- Default: false
- IRC_DIRECTION (OPTKING)
OPTKING — IRC mapping direction
- Type: string
- Possible Values: FORWARD, BACKWARD
- Default: FORWARD
- IRC_STEP_SIZE (OPTKING)
OPTKING — IRC step size in bohr(amu)
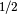 .
.- Type: double
- Default: 0.2
- IRC_STOP (OPTKING)
OPTKING — Decide when to stop IRC calculations
- Type: string
- Possible Values: ASK, STOP, GO
- Default: STOP
- ISTOP (DETCI)
DETCI — Do stop DETCI after string information is formed and before integrals are read?
- Type: boolean
- Default: false
- IVO (TRANSQT)
TRANSQT — Do form improved virtual orbitals (IVO)?
- Type: boolean
- Default: false
- J_FILE (TRANSQT)
TRANSQT — Half-transformed integrals
- Type: integer
- Default: 91
- JOBTYPE (CCLAMBDA)
CCLAMBDA (Expert) — Type of job being performed
- Type: string
- Default: No Default
- KEEP_INTCOS (OPTKING)
OPTKING — Keep internal coordinate definition file.
- Type: boolean
- Default: false
- KEEP_J (TRANSQT)
TRANSQT — Do keep half-transformed integrals?
- Type: boolean
- Default: false
- KEEP_OEIFILE (CCSORT)
CCSORT — Do retain the input one-electron integrals?
- Type: boolean
- Default: false
- KEEP_PRESORT (TRANSQT)
TRANSQT — Do keep presort file?
- Type: boolean
- Default: false
- KEEP_TEIFILE (CCSORT)
CCSORT — Do retain the input two-electron integrals?
- Type: boolean
- Default: false
- LAG_IN_FILE (TRANSQT)
TRANSQT — MO-basis MO-lagrangian file
- Type: integer
- Default: PSIF_MO_LAG
- LAGRAN_DOUBLE (TRANSQT)
TRANSQT — Do multiply the MO-lagrangian by 2.0?
- Type: boolean
- Default: false
- LAGRAN_HALVE (TRANSQT)
TRANSQT — Do divide the MO-lagrangian by 2.0?
- Type: boolean
- Default: false
- LAMBDA_MAXITER (DCFT)
DCFT — Maximum number of density cumulant update micro-iterations per macro-iteration (for ALOGRITHM = TWOSTEP). Same keyword controls the maximum number of density cumulant response micro-iterations per macro-iteration for the solution of the response equations (for RESPONSE_ALOGRITHM = TWOSTEP)
- Type: integer
- Default: 50
- LEVEL_SHIFT (MCSCF)
MCSCF — Level shift to aid convergence
- Type: double
- Default: 0.0
- LEVEL_SHIFT (OCC)
OCC — Level shift to aid convergence
- Type: double
- Default: 0.02
- LINEAR (CCRESPONSE)
CCRESPONSE — Do Bartlett size-extensive linear model?
- Type: boolean
- Default: false
- LINEQ_SOLVER (OCC)
OCC — The solver will be used for simultaneous linear equations.
- Type: string
- Possible Values: CDGESV, FLIN, POPLE
- Default: CDGESV
- LINESEARCH_STATIC_MAX (OPTKING)
OPTKING — If doing a static line search, this fixes the largest step, whose largest change in an internal coordinate is set to this value (in au)
- Type: double
- Default: 0.100
- LINESEARCH_STATIC_MIN (OPTKING)
OPTKING — If doing a static line search, this fixes the shortest step, whose largest change in an internal coordinate is set to this value (in au)
- Type: double
- Default: 0.001
- LINESEARCH_STATIC_N (OPTKING)
OPTKING — If doing a static line search, scan this many points.
- Type: integer
- Default: 8
- LOCAL (CCENERGY)
CCENERGY — Do simulate the effects of local correlation techniques?
- Type: boolean
- Default: false
- LOCAL (CCEOM)
CCEOM — Do simulate the effects of local correlation techniques?
- Type: boolean
- Default: false
- LOCAL (CCLAMBDA)
CCLAMBDA — Do simulate the effects of local correlation techniques?
- Type: boolean
- Default: false
- LOCAL (CCRESPONSE)
CCRESPONSE — Do simulate local correlation?
- Type: boolean
- Default: false
- LOCAL (CCSORT)
CCSORT — Do simulate the effects of local correlation techniques?
- Type: boolean
- Default: false
- LOCAL (CIS)
CIS — Do simulate the effects of local correlation techniques?
- Type: boolean
- Default: false
- LOCAL_AMPS_PRINT_CUTOFF (CIS)
CIS — Cutoff value for printing local amplitudes
- Type: double
- Default: 0.60
- LOCAL_CORE_CUTOFF (CCSORT)
CCSORT — Local core cutoff value
- Type: double
- Default: 0.05
- LOCAL_CPHF_CUTOFF (CCENERGY)
CCENERGY — Cutoff value for local-coupled-perturbed-Hartree-Fock
- Type: double
- Default: 0.10
- LOCAL_CPHF_CUTOFF (CCLAMBDA)
CCLAMBDA — Cutoff value for local-coupled-perturbed-Hartree-Fock
- Type: double
- Default: 0.10
- LOCAL_CPHF_CUTOFF (CCRESPONSE)
CCRESPONSE — Cutoff value for local-coupled-perturbed-Hartree-Fock
- Type: double
- Default: 0.10
- LOCAL_CPHF_CUTOFF (CCSORT)
CCSORT — Cutoff value for local-coupled-perturbed-Hartree-Fock
- Type: double
- Default: 0.10
- LOCAL_CUTOFF (CCENERGY)
CCENERGY — Value (always between one and zero) for the Broughton-Pulay completeness check used to contruct orbital domains for local-CC calculations. See J. Broughton and P. Pulay, J. Comp. Chem. 14, 736-740 (1993) and C. Hampel and H.-J. Werner, J. Chem. Phys. 104, 6286-6297 (1996).
- Type: double
- Default: 0.02
- LOCAL_CUTOFF (CCEOM)
CCEOM — Value (always between one and zero) for the Broughton-Pulay completeness check used to contruct orbital domains for local-CC calculations. See J. Broughton and P. Pulay, J. Comp. Chem. 14, 736-740 (1993) and C. Hampel and H.-J. Werner, J. Chem. Phys. 104, 6286-6297 (1996).
- Type: double
- Default: 0.02
- LOCAL_CUTOFF (CCLAMBDA)
CCLAMBDA — Value (always between one and zero) for the Broughton-Pulay completeness check used to contruct orbital domains for local-CC calculations. See J. Broughton and P. Pulay, J. Comp. Chem. 14, 736-740 (1993) and C. Hampel and H.-J. Werner, J. Chem. Phys. 104, 6286-6297 (1996).
- Type: double
- Default: 0.02
- LOCAL_CUTOFF (CCRESPONSE)
CCRESPONSE — Value (always between one and zero) for the Broughton-Pulay completeness check used to contruct orbital domains for local-CC calculations. See J. Broughton and P. Pulay, J. Comp. Chem. 14, 736-740 (1993) and C. Hampel and H.-J. Werner, J. Chem. Phys. 104, 6286-6297 (1996).
- Type: double
- Default: 0.01
- LOCAL_CUTOFF (CCSORT)
CCSORT — Value (always between one and zero) for the Broughton-Pulay completeness check used to contruct orbital domains for local-CC calculations. See J. Broughton and P. Pulay, J. Comp. Chem. 14, 736-740 (1993) and C. Hampel and H.-J. Werner, J. Chem. Phys. 104, 6286-6297 (1996).
- Type: double
- Default: 0.02
- LOCAL_CUTOFF (CIS)
CIS — Value (always between one and zero) for the Broughton-Pulay completeness check used to contruct orbital domains for local-CC calculations. See J. Broughton and P. Pulay, J. Comp. Chem. 14, 736-740 (1993) and C. Hampel and H.-J. Werner, J. Chem. Phys. 104, 6286-6297 (1996).
- Type: double
- Default: 0.02
- LOCAL_CUTOFF (LMP2)
LMP2 — Localization cutoff
- Type: double
- Default: 0.02
- LOCAL_DO_SINGLES (CCEOM)
CCEOM —
- Type: boolean
- Default: true
- LOCAL_DOMAIN_MAG (CCSORT)
CCSORT — Do generate magnetic-field CPHF solutions for local-CC?
- Type: boolean
- Default: false
- LOCAL_DOMAIN_POLAR (CCSORT)
CCSORT — Do use augment domains with polarized orbitals?
- Type: boolean
- Default: false
- LOCAL_DOMAIN_SEP (CCSORT)
CCSORT —
- Type: boolean
- Default: false
- LOCAL_FILTER_SINGLES (CCEOM)
CCEOM — Do apply local filtering to singles amplitudes?
- Type: boolean
- Default: true
- LOCAL_FILTER_SINGLES (CCLAMBDA)
CCLAMBDA — Do apply local filtering to single de-excitation (
 amplitudes?
amplitudes?- Type: boolean
- Default: true
- LOCAL_FILTER_SINGLES (CCRESPONSE)
CCRESPONSE — Do apply local filtering to single excitation amplitudes?
- Type: boolean
- Default: false
- LOCAL_FILTER_SINGLES (CCSORT)
CCSORT — Do apply local filtering to single excitation amplitudes?
- Type: boolean
- Default: false
- LOCAL_GHOST (CCEOM)
CCEOM — Permit ghost atoms to hold projected atomic orbitals to include in the virtual space in local-EOM-CCSD calculations
- Type: integer
- Default: -1
- LOCAL_GHOST (CIS)
CIS —
- Type: integer
- Default: -1
- LOCAL_METHOD (CCENERGY)
CCENERGY — Type of local-CCSD scheme to be simulated. WERNER selects the method developed by H.-J. Werner and co-workers, and AOBASIS selects the method developed by G.E. Scuseria and co-workers (currently inoperative).
- Type: string
- Possible Values: WERNER, AOBASIS
- Default: WERNER
- LOCAL_METHOD (CCEOM)
CCEOM — Type of local-CCSD scheme to be simulated. WERNER selects the method developed by H.-J. Werner and co-workers, and AOBASIS selects the method developed by G.E. Scuseria and co-workers (currently inoperative).
- Type: string
- Possible Values: WERNER, AOBASIS
- Default: WERNER
- LOCAL_METHOD (CCLAMBDA)
CCLAMBDA — Type of local-CCSD scheme to be simulated. WERNER (unique avaliable option) selects the method developed by H.-J. Werner and co-workers.
- Type: string
- Default: WERNER
- LOCAL_METHOD (CCRESPONSE)
CCRESPONSE — Type of local-CCSD scheme to be simulated. WERNER (unique avaliable option) selects the method developed by H.-J. Werner and co-workers.
- Type: string
- Default: WERNER
- LOCAL_METHOD (CCSORT)
CCSORT — Type of local-CCSD scheme to be simulated. WERNER (unique avaliable option) selects the method developed by H.-J. Werner and co-workers.
- Type: string
- Default: WERNER
- LOCAL_METHOD (CIS)
CIS — Type of local-CIS scheme to be simulated. WERNER selects the method developed by H.-J. Werner and co-workers, and AOBASIS selects the method developed by G.E. Scuseria and co-workers.
- Type: string
- Possible Values: AOBASIS, WERNER
- Default: WERNER
- LOCAL_PAIRDEF (CCENERGY)
CCENERGY — Definition of local pair domains, default is BP, Boughton-Pulay.
- Type: string
- Possible Values: BP, RESPONSE
- Default: BP
- LOCAL_PAIRDEF (CCLAMBDA)
CCLAMBDA — Definition of local pair domains
- Type: string
- Default: No Default
- LOCAL_PAIRDEF (CCRESPONSE)
CCRESPONSE — Definition of local pair domains
- Type: string
- Default: NONE
- LOCAL_PAIRDEF (CCSORT)
CCSORT — Definition of local pair domains, unique avaliable option is BP, Boughton-Pulay.
- Type: string
- Default: BP
- LOCAL_PRECONDITIONER (CCEOM)
CCEOM — Preconditioner will be used in local CC computations
- Type: string
- Possible Values: HBAR, FOCK
- Default: HBAR
- LOCAL_WEAKP (CCENERGY)
CCENERGY — Desired treatment of “weak pairs” in the local-CCSD method. A value of NEGLECT ignores weak pairs entirely. A value of NONE treats weak pairs in the same manner as strong pairs. A value of MP2 uses second-order perturbation theory to correct the local-CCSD energy computed with weak pairs ignored.
- Type: string
- Possible Values: NONE, NEGLECT, MP2
- Default: NONE
- LOCAL_WEAKP (CCEOM)
CCEOM — Desired treatment of “weak pairs” in the local-CCSD method. A value of NEGLECT ignores weak pairs entirely. A value of NONE treats weak pairs in the same manner as strong pairs. A value of MP2 uses second-order perturbation theory to correct the local-CCSD energy computed with weak pairs ignored.
- Type: string
- Possible Values: NONE, MP2, NEGLECT
- Default: NONE
- LOCAL_WEAKP (CCLAMBDA)
CCLAMBDA — Desired treatment of “weak pairs” in the local-CCSD method. The value of NONE (unique avaliable option) treats weak pairs in the same manner as strong pairs.
- Type: string
- Default: NONE
- LOCAL_WEAKP (CCRESPONSE)
CCRESPONSE — Desired treatment of “weak pairs” in the local-CCSD method. The value of NONE (unique avaliable option) treats weak pairs in the same manner as strong pairs.
- Type: string
- Default: NONE
- LOCAL_WEAKP (CCSORT)
CCSORT — Desired treatment of “weak pairs” in the local-CCSD method. The value of NONE (unique avaliable option) treats weak pairs in the same manner as strong pairs.
- Type: string
- Default: NONE
- LOCAL_WEAKP (CIS)
CIS — Desired treatment of “weak pairs” in the local-CIS method. A value of NEGLECT ignores weak pairs entirely. A value of NONE treats weak pairs in the same manner as strong pairs. A value of MP2 uses second-order perturbation theory to correct the local-CIS energy computed with weak pairs ignored.
- Type: string
- Possible Values: MP2, NEGLECT, NONE
- Default: MP2
- LOCK_OCC (DCFT)
DCFT (Expert) — Controls whether to force the occupation to be that of the SCF guess. For practical applications only the default must be used
- Type: boolean
- Default: true
- LOCK_SINGLET (PSIMRCC)
PSIMRCC — Do lock onto a singlet root?
- Type: boolean
- Default: false
- LSE (DETCI)
DETCI — Do use least-squares extrapolation in iterative solution of CI vector?
- Type: boolean
- Default: false
- LSE_COLLAPSE (DETCI)
DETCI — Number of iterations between least-squares extrapolations
- Type: integer
- Default: 3
- LSE_TOLERANCE (DETCI)
DETCI — Minimum converged energy for least-squares extrapolation to be performed
- Type: conv double
- Default: 3
- M_FILE (TRANSQT)
TRANSQT — Output integrals file
- Type: integer
- Default: 0
- MADMP2_SLEEP (DFMP2)
DFMP2 (Expert) — A helpful option, used only in debugging the MADNESS version
- Type: integer
- Default: 0
- MAT_NUM_COLUMN_PRINT (GLOBALS)
GLOBALS (Expert) — Number of columns to print in calls to Matrix::print_mat.
- Type: integer
- Default: 5
- MAX_BUCKETS (TRANSQT)
TRANSQT — Maximum buckets
- Type: integer
- Default: 499
- MAX_CCD_DIISVECS (SAPT)
SAPT — Maximum number of vectors used in CCD-DIIS
- Type: integer
- Default: 10
- MAX_DISP_G_CONVERGENCE (OPTKING)
OPTKING — Convergence criterion for geometry optmization: maximum displacement (internal coordinates, atomic units).
- Type: conv double
- Default: 1.2e-3
- MAX_ENERGY_G_CONVERGENCE (OPTKING)
OPTKING — Convergence criterion for geometry optmization: maximum energy change.
- Type: conv double
- Default: 1.0e-6
- MAX_FORCE_G_CONVERGENCE (OPTKING)
OPTKING — Convergence criterion for geometry optmization: maximum force (internal coordinates, atomic units).
- Type: conv double
- Default: 3.0e-4
- MAX_MOGRAD_CONVERGENCE (OCC)
OCC — Convergence criterion for maximum orbital gradient
- Type: conv double
- Default: 1e-3
- MAX_NUM_VECS (DETCI)
DETCI — Maximum number of Davidson subspace vectors which can be held on disk for the CI coefficient and sigma vectors. (There is one H(diag) vector and the number of D vectors is equal to the number of roots). When the number of vectors on disk reaches the value of MAX_NUM_VECS, the Davidson subspace will be collapsed to COLLAPSE_SIZE vectors for each root. This is very helpful for saving disk space. Defaults to MAXITER * NUM_ROOTS + NUM_INIT_VECS.
- Type: integer
- Default: 0
- MAXITER (CCENERGY)
CCENERGY — Maximum number of iterations to solve the CC equations
- Type: integer
- Default: 50
- MAXITER (CCEOM)
CCEOM — Maximum number of iterations
- Type: integer
- Default: 80
- MAXITER (CCLAMBDA)
CCLAMBDA — Maximum number of iterations
- Type: integer
- Default: 50
- MAXITER (CCRESPONSE)
CCRESPONSE — Maximum number of iterations to converge perturbed amplitude equations
- Type: integer
- Default: 50
- MAXITER (CIS)
CIS — Maximum number of iterations
- Type: integer
- Default: 500
- MAXITER (DCFT)
DCFT — Maximum number of the macro-iterations for both the energy and the solution of the response equations
- Type: integer
- Default: 40
- MAXITER (DETCI)
DETCI — Maximum number of iterations to diagonalize the Hamiltonian
- Type: integer
- Default: 12
- MAXITER (DFTSAPT)
DFTSAPT — The maximum number of iterations in CPKS
- Type: integer
- Default: 100
- MAXITER (FNOCC)
FNOCC — Maximum number of CC iterations
- Type: integer
- Default: 100
- MAXITER (LMP2)
LMP2 — Maximum number of iterations
- Type: integer
- Default: 50
- MAXITER (MCSCF)
MCSCF — Maximum number of iterations
- Type: integer
- Default: 100
- MAXITER (PSIMRCC)
PSIMRCC — Maximum number of iterations to determine the amplitudes
- Type: integer
- Default: 100
- MAXITER (SAPT)
SAPT — Maxmum number of CPHF iterations
- Type: integer
- Default: 50
- MAXITER (SCF)
SCF — Maximum number of iterations
- Type: integer
- Default: 100
- MEMORY (ADC)
ADC — The amount of memory available (in Mb)
- Type: integer
- Default: 1000
- MEMORY (LMP2)
LMP2 — The amount of memory available (in Mb)
- Type: integer
- Default: 2000
- MIN_CCD_DIISVECS (SAPT)
SAPT — Minimumnumber of vectors used in CCD-DIIS
- Type: integer
- Default: 4
- MIXED (DETCI)
DETCI (Expert) — Do allow “mixed” RAS II/RAS III excitations into the CI space? If FALSE, then if there are any electrons in RAS III, then the number of holes in RAS I cannot exceed the given excitation level EX_LEVEL.
- Type: boolean
- Default: true
- MIXED4 (DETCI)
DETCI (Expert) — Do allow “mixed” excitations involving RAS IV into the CI space. Useful to specify a split-virtual CISD[TQ] computation. If FALSE, then if there are any electrons in RAS IV, then the number of holes in RAS I cannot exceed the given excitation level EX_LEVEL.
- Type: boolean
- Default: true
- MO_DIIS_NUM_VECS (OCC)
OCC — Number of vectors used in orbital DIIS
- Type: integer
- Default: 6
- MO_MAXITER (OCC)
OCC — Maximum number of iterations to determine the orbitals
- Type: integer
- Default: 50
- MO_READ (MCSCF)
MCSCF — Do read in from file the MOs from a previous computation?
- Type: boolean
- Default: true
- MO_READ (OCC)
OCC — Do read coefficient matrices from external files of a previous OMP2 or OMP3 computation?
- Type: boolean
- Default: false
- MO_RELAX (DCFT)
DCFT (Expert) — Controls whether to relax the orbitals during the energy computation or not (for debug puproses only). For practical applications only the default must be used
- Type: boolean
- Default: true
- MO_STEP_MAX (OCC)
OCC — Maximum step size in orbital-optimization procedure
- Type: double
- Default: 0.5
- MO_WRITE (OCC)
OCC — Do write coefficient matrices to external files for direct reading MOs in a subsequent job?
- Type: boolean
- Default: false
- MODE (TRANSQT)
TRANSQT — The way of transformation, from ao basis to mo basis or vice versa
- Type: string
- Possible Values: TO_MO, TO_AO
- Default: TO_MO
- MODULE (CPHF)
CPHF — What app to test?
- Type: string
- Possible Values: RCIS, RCPHF, RTDHF, RCPKS, RTDA, RTDDFT
- Default: RCIS
- MOGRAD_DAMPING (OCC)
OCC — Damping factor for the orbital gradient (Rendell et al., JCP, vol. 87, pp. 5976, 1987)
- Type: double
- Default: 1.0
- MOLDEN_WRITE (SCF)
SCF — Do write a MOLDEN output file? If so, the filename will end in .molden, and the prefix is determined by WRITER_FILE_LABEL (if set), or else by the name of the output file plus the name of the current molecule.
- Type: boolean
- Default: false
- MOM_OCC (SCF)
SCF — The absolute indices of orbitals to excite from in MOM (+/- for alpha/beta)
- Type: array
- Default: No Default
- MOM_START (SCF)
SCF — The iteration to start MOM on (or 0 for no MOM)
- Type: integer
- Default: 0
- MOM_VIR (SCF)
SCF — The absolute indices of orbitals to excite to in MOM (+/- for alpha/beta)
- Type: array
- Default: No Default
- MOORDER (TRANSQT)
TRANSQT — Numbering of MOs for reordering requests?
- Type: array
- Default: No Default
- MP2_AMPS_PRINT (CCENERGY)
CCENERGY — Do print the MP2 amplitudes which are the starting guesses for RHF and UHF reference functions?
- Type: boolean
- Default: false
- MP2_CCSD_METHOD (PSIMRCC)
PSIMRCC — How to perform MP2_CCSD computations
- Type: string
- Possible Values: I, IA, II
- Default: II
- MP2_GUESS (PSIMRCC)
PSIMRCC — Do start from a MP2 guess?
- Type: boolean
- Default: true
- MP2_OS_SCALE (CCENERGY)
CCENERGY — MP2 opposite-spin scaling value
- Type: double
- Default: 1.20
- MP2_OS_SCALE (DFMP2)
DFMP2 — OS Scale
- Type: double
- Default: 6.0/5.0
- MP2_OS_SCALE (LMP2)
LMP2 — The scale factor used for opposite-spin pairs in SCS computations
- Type: double
- Default: 6.0/5.0
- MP2_OS_SCALE (OCC)
OCC — MP2 opposite-spin scaling value
- Type: double
- Default: 6.0/5.0
- MP2_SCALE_OS (FNOCC)
FNOCC — Opposite-spin scaling factor for SCS-MP2
- Type: double
- Default: 1.20
- MP2_SCALE_SS (FNOCC)
FNOCC — Same-spin scaling factor for SCS-MP2
- Type: double
- Default: 1.0/3.0
- MP2_SOS_SCALE (OCC)
OCC — MP2 Spin-opposite scaling (SOS) value
- Type: double
- Default: 1.3
- MP2_SOS_SCALE2 (OCC)
OCC — Spin-opposite scaling (SOS) value for optimized-MP2 orbitals
- Type: double
- Default: 1.2
- MP2_SS_SCALE (CCENERGY)
CCENERGY — MP2 same-spin scaling value
- Type: double
- Default: 1.0/3.0
- MP2_SS_SCALE (DFMP2)
DFMP2 — SS Scale
- Type: double
- Default: 1.0/3.0
- MP2_SS_SCALE (LMP2)
LMP2 — The scale factor used for same-spin pairs in SCS computations
- Type: double
- Default: 1.0/3.0
- MP2_SS_SCALE (OCC)
OCC — MP2 same-spin scaling value
- Type: double
- Default: 1.0/3.0
- MP2_TYPE (DFMP2)
DFMP2 — Algorithm to use for the MP2 computation
- Type: string
- Possible Values: DF, CONV
- Default: DF
- MP2_TYPE (OCC)
OCC — Algorithm to use for non-OO MP2 computation
- Type: string
- Possible Values: DF, CONV
- Default: DF
- MP2R12A (TRANSQT)
TRANSQT — Transformations for explicitly-correlated MP2 methods
- Type: string
- Possible Values: MP2R12AERI, MP2R12AR12, MP2R12AR12T1
- Default: MP2R12AERI
- MPN (DETCI)
DETCI — Do compute the MPn series out to kth order where k is determined by MAX_NUM_VECS ? For open-shell systems (REFERENCE is ROHF, WFN is ZAPTN), DETCI will compute the ZAPTn series. GUESS_VECTOR must be set to UNIT, HD_OTF must be set to TRUE, and HD_AVG must be set to orb_ener; these should happen by default for MPN = TRUE.
- Type: boolean
- Default: false
- MPN_ORDER_SAVE (DETCI)
DETCI (Expert) — If 0, save the MPn energy; if 1, save the MP(2n-1) energy (if available from MPN_WIGNER = true); if 2, save the MP(2n-2) energy (if available from MPN_WIGNER = true).
- Type: integer
- Default: 0
- MPN_SCHMIDT (DETCI)
DETCI (Expert) — Do employ an orthonormal vector space rather than storing the kth order wavefunction?
- Type: boolean
- Default: false
- MPN_WIGNER (DETCI)
DETCI (Expert) — Do use Wigner formulas in the
 series?
series?- Type: boolean
- Default: true
- MRCC_LEVEL (MRCC)
MRCC — Maximum excitation level. This is used ONLY if it is explicity set by the user. Single-reference case: all excitations up to this level are included, e.g., 2 for CCSD, 3 for CCSDT, 4 for CCSDTQ, etc. This becomes ex.lev (option #1) in fort.56.
- Type: integer
- Default: 2
- MRCC_METHOD (MRCC)
MRCC (Expert) — If more than one root is requested and calc=1, LR-CC (EOM-CC) calculation is performed automatically for the excited states. This overrides all automatic determination of method and will only work with energy(). This becomes CC/CI (option #5) in fort.56 .. table:: MRCC methods +——-+————–+————————————————————-+ + Value + Method + Description + +=======+==============+=============================================================+ + 1 + CC + + +——-+————–+————————————————————-+ + 2 + CC(n-1)[n] + + +——-+————–+————————————————————-+ + 3 + CC(n-1)(n) + (CC(n-1)[n] energy is also calculated) + +——-+————–+————————————————————-+ + 4 + CC(n-1)(n)_L + (CC(n-1)[n] and CC(n-1)(n) energies are also calculated) + +——-+————–+————————————————————-+ + 5 + CC(n)-1a + + +——-+————–+————————————————————-+ + 6 + CC(n)-1b + + +——-+————–+————————————————————-+ + 7 + CCn + + +——-+————–+————————————————————-+ + 8 + CC(n)-3 + + +——-+————–+————————————————————-+
- Type: integer
- Default: 1
- MRCC_NUM_SINGLET_ROOTS (MRCC)
MRCC — Number of singlet roots. (Strictly speaking number of of roots with M_s=0 and S is even.) Use this option only with closed shell reference determinant, it must be zero otherwise. This becomes nsing (option #2) in fort.56.
- Type: integer
- Default: 1
- MRCC_NUM_TRIPLET_ROOTS (MRCC)
MRCC — Number of triplet roots. (Strictly speaking number of of roots with
 and S is odd.) See notes at option MRCC_NUM_SINGLET_ROOTS. This becomes ntrip (option #3) in fort.56.
and S is odd.) See notes at option MRCC_NUM_SINGLET_ROOTS. This becomes ntrip (option #3) in fort.56.- Type: integer
- Default: 0
- MRCC_OMP_NUM_THREADS (MRCC)
MRCC (Expert) — Sets the OMP_NUM_THREADS environment variable before calling MRCC. If the environment variable OMP_NUM_THREADS is set prior to calling PSI4 then that value is used. When set, this option overrides everything. Be aware the -n command-line option described in section Threading does not affect MRCC.
- Type: integer
- Default: 1
- MRCC_RESTART (MRCC)
MRCC (Expert) — The program restarts from the previously calculated parameters if it is 1. In case it is 2, the program executes automatically the lower-level calculations of the same type consecutively (e.g., CCSD, CCSDT, and CCSDTQ if CCSDTQ is requested) and restarts each calculation from the previous one (rest=2 is available only for energy calculations). Currently, only a value of 0 and 2 are supported. This becomes rest (option #4) in fort.56.
- Type: integer
- Default: 0
- MS0 (DETCI)
DETCI — Do use the
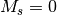 component of the state? Defaults to TRUE if closed-shell and FALSE otherwise. Related to the S option.
component of the state? Defaults to TRUE if closed-shell and FALSE otherwise. Related to the S option.- Type: boolean
- Default: false
- NAT_ORBS (FNOCC)
FNOCC — Do use MP2 NOs to truncate virtual space for QCISD/CCSD and (T)?
- Type: boolean
- Default: false
- NAT_ORBS (OCC)
OCC — Do compute natural orbitals?
- Type: boolean
- Default: false
- NAT_ORBS (SAPT)
SAPT — Do natural orbitals to speed up evaluation of the triples contribution to dispersion by truncating the virtual orbital space? Recommended true for all SAPT computations.
- Type: boolean
- Default: false
- NAT_ORBS_T2 (SAPT)
SAPT — Do use MP2 natural orbital approximations for the
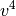 block of two-electron integrals in the evaluation of second-order T2 amplitudes? This approximation is promising for accuracy and computational savings, but it has not been rigorously tested.
block of two-electron integrals in the evaluation of second-order T2 amplitudes? This approximation is promising for accuracy and computational savings, but it has not been rigorously tested.- Type: boolean
- Default: false
- NAT_ORBS_WRITE (DETCI)
DETCI — Do write the natural orbitals?
- Type: boolean
- Default: false
- NAT_ORBS_WRITE_ROOT (DETCI)
DETCI — Sets the root number for which CI natural orbitals are written to PSIF_CHKPT. The default value is 1 (lowest root).
- Type: integer
- Default: 1
- NEGLECT_DISTANT_PAIR (LMP2)
LMP2 — Do neglect distant pairs?
- Type: boolean
- Default: true
- NEW_TRIPLES (CCENERGY)
CCENERGY — Do use new triples?
- Type: boolean
- Default: true
- NEW_TRIPLES (CCEOM)
CCEOM — Do use new triples?
- Type: boolean
- Default: true
- NEWTON_CONVERGENCE (ADC)
ADC — The convergence criterion for pole searching step.
- Type: conv double
- Default: 1e-7
- NO_DFILE (DETCI)
DETCI (Expert) — Do use the last vector space in the BVEC file to write scratch DVEC rather than using a separate DVEC file? (Only possible if NUM_ROOTS = 1.)
- Type: boolean
- Default: false
- NO_RESPONSE (SAPT)
SAPT — Don’t solve the CPHF equations? Evaluate
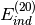 and
and 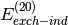 instead of their response-including coupterparts. Only turn on this option if the induction energy is not going to be used.
instead of their response-including coupterparts. Only turn on this option if the induction energy is not going to be used.- Type: boolean
- Default: false
- NO_SINGLES (PSIMRCC)
PSIMRCC — Do disregard updating single excitation amplitudes?
- Type: boolean
- Default: false
- NORM_TOLERANCE (ADC)
ADC — The cutoff norm of residual vector in SEM step.
- Type: conv double
- Default: 1e-6
- NUM_AMPS_PRINT (ADC)
ADC — Number of components of transition amplitudes printed
- Type: integer
- Default: 5
- NUM_AMPS_PRINT (CCENERGY)
CCENERGY — Number of important
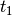 and
and 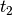 amplitudes to print
amplitudes to print- Type: integer
- Default: 10
- NUM_AMPS_PRINT (CCEOM)
CCEOM — Number of important CC amplitudes to print
- Type: integer
- Default: 5
- NUM_AMPS_PRINT (CCLAMBDA)
CCLAMBDA — Number of important CC amplitudes per excitation level to print. CC analog to NUM_DETS_PRINT.
- Type: integer
- Default: 10
- NUM_AMPS_PRINT (CCRESPONSE)
CCRESPONSE — Number of important CC amplitudes per excitation level to print. CC analog to NUM_DETS_PRINT.
- Type: integer
- Default: 5
- NUM_AMPS_PRINT (DETCI)
DETCI — Number of important CC amplitudes per excitation level to print. CC analog to NUM_DETS_PRINT.
- Type: integer
- Default: 10
- NUM_DETS_PRINT (DETCI)
DETCI — Number of important determinants to print
- Type: integer
- Default: 20
- NUM_FROZEN_DOCC (GLOBALS)
GLOBALS — The number of core orbitals to freeze in later correlated computations. This trumps FREEZE_CORE.
- Type: integer
- Default: 0
- NUM_FROZEN_UOCC (GLOBALS)
GLOBALS — The number of virtual orbitals to freeze in later correlated computations.
- Type: integer
- Default: 0
- NUM_INIT_VECS (DETCI)
DETCI (Expert) — The number of initial vectors to use in the CI iterative procedure. Defaults to the number of roots.
- Type: integer
- Default: 0
- NUM_ROOTS (DETCI)
DETCI — number of CI roots to find
- Type: integer
- Default: 1
- NUM_VECS_PRINT (STABILITY)
STABILITY — Number of lowest MO Hessian eigenvalues to print
- Type: integer
- Default: 0
- NUM_VECS_WRITE (DETCI)
DETCI — Number of vectors to export
- Type: integer
- Default: 1
- OCC_ORBS_PRINT (OCC)
OCC — Do print OCC orbital energies?
- Type: boolean
- Default: false
- OCC_TOLERANCE (FNOCC)
FNOCC — Cutoff for occupation of MP2 NO orbitals in FNO-QCISD/CCSD(T) ( only valid if NAT_ORBS = true )
- Type: conv double
- Default: 1.0e-6
- OCC_TOLERANCE (SAPT)
SAPT — Minimum occupation (eigenvalues of the MP2 OPDM) below which virtual natural orbitals are discarded for evaluating the triples contribution to dispersion.
- Type: conv double
- Default: 1.0e-6
- OEI_A_FILE (TRANSQT)
TRANSQT — Alpha-spin one-electron parameters file
- Type: integer
- Default: PSIF_OEI
- OEI_B_FILE (TRANSQT)
TRANSQT — Beta-spin one-electron parameters file
- Type: integer
- Default: PSIF_OEI
- OEI_FILE (TRANSQT)
TRANSQT — One-electron parameters file
- Type: integer
- Default: PSIF_OEI
- OFFDIAGONAL_CCSD_T (PSIMRCC)
PSIMRCC — Do include the off-diagonal corrections in (T) computations?
- Type: boolean
- Default: true
- OMEGA (CCRESPONSE)
CCRESPONSE — Array that specifies the desired frequencies of the incident radiation field in CCLR calculations. If only one element is given, the units will be assumed to be atomic units. If more than one element is given, then the units must be specified as the final element of the array. Acceptable units are HZ, NM, EV, and AU.
- Type: array
- Default: No Default
- OMEGA (CCSORT)
CCSORT — Energy of applied field [au] for dynamic properties
- Type: array
- Default: No Default
- OMEGA (RESPONSE)
RESPONSE — Array that specifies the desired frequencies of the incident radiation field in CCLR calculations. If only one element is given, the units will be assumed to be atomic units. If more than one element is given, then the units must be specified as the final element of the array. Acceptable units are HZ, NM, EV, and AU.
- Type: array
- Default: No Default
- OMEGA_ERF (MINTS)
MINTS — Omega scaling for Erf and Erfc.
- Type: double
- Default: 0.20
- OMP_N_THREAD (CPHF)
CPHF — The maximum number of integral threads (0 for omp_get_max_threads())
- Type: integer
- Default: 0
- ONEPDM (CCDENSITY)
CCDENSITY — Do compute one-particle density matrix?
- Type: boolean
- Default: false
- ONEPDM (DFMP2)
DFMP2 — Do compute one-particle density matrix?
- Type: boolean
- Default: false
- ONEPDM_GRID_CUTOFF (CCDENSITY)
CCDENSITY — Cutoff (e/A^3) for printing one-particle density matrix values on a grid
- Type: double
- Default: 1.0e-30
- ONEPDM_GRID_DUMP (CCDENSITY)
CCDENSITY — Write one-particle density matrix on a grid to file opdm.dx
- Type: boolean
- Default: false
- ONEPDM_GRID_STEPSIZE (CCDENSITY)
CCDENSITY — Stepsize (Angstrom) for one-particle density matrix values on a grid
- Type: double
- Default: 0.1
- ONEPOT_GRID_READ (SCF)
SCF — Read an external potential from the .dx file?
- Type: boolean
- Default: false
- OPDM (DETCI)
DETCI — Do compute one-particle density matrix if not otherwise required?
- Type: boolean
- Default: false
- OPDM_AVG (DETCI)
DETCI — Do average the OPDM over several roots in order to obtain a state-average one-particle density matrix? This density matrix can be diagonalized to obtain the CI natural orbitals.
- Type: boolean
- Default: false
- OPDM_IN_FILE (TRANSQT)
TRANSQT — MO-basis one-particle density matrix file
- Type: integer
- Default: PSIF_MO_OPDM
- OPDM_KE (DETCI)
DETCI (Expert) — Do compute the kinetic energy contribution from the correlated part of the one-particle density matrix?
- Type: boolean
- Default: false
- OPDM_OUT_FILE (TRANSQT)
TRANSQT — AO-basis one-particle density matrix file
- Type: integer
- Default: PSIF_AO_OPDM
- OPDM_PRINT (DETCI)
DETCI — Do print the one-particle density matrix for each root?
- Type: boolean
- Default: false
- OPDM_RELAX (CCDENSITY)
CCDENSITY — Do relax the one-particle density matrix?
- Type: boolean
- Default: false
- OPDM_RELAX (DFMP2)
DFMP2 — Do relax the one-particle density matrix?
- Type: boolean
- Default: true
- OPT_METHOD (OCC)
OCC — The optimization algorithm. Modified Steepest-Descent (MSD) takes a Newton-Raphson (NR) step with a crude approximation to diagonal elements of the MO Hessian. The ORB_RESP option obtains the orbital rotation parameters by solving the orbital-reponse (coupled-perturbed CC) equations. Additionally, for both methods a DIIS extrapolation will be performed with the DO_DIIS = TRUE option.
- Type: string
- Possible Values: MSD, ORB_RESP
- Default: ORB_RESP
- OPT_TYPE (OPTKING)
OPTKING — Specifies minimum search, transition-state search, or IRC following
- Type: string
- Possible Values: MIN, TS, IRC
- Default: MIN
- ORB_OPT (OCC)
OCC — Do optimize the orbitals?
- Type: boolean
- Default: true
- ORB_RESP_SOLVER (OCC)
OCC — The algorithm will be used for solving the orbital-response equations. The LINEQ option create the MO Hessian and solve the simultaneous linear equations with method choosen by the LINEQ_SOLVER option. The PCG option does not create the MO Hessian explicitly, instead it solves the simultaneous equations iteratively with the preconditioned conjugate gradient method.
- Type: string
- Possible Values: PCG, LINEQ
- Default: PCG
- ORTH_TYPE (OCC)
OCC — The algorithm for orthogonalization of MOs
- Type: string
- Possible Values: GS, MGS
- Default: MGS
- P (THERMO)
THERMO — Pressure in Pascal for thermodynamic analysis.
- Type: double
- Default: 101325
- PAIR_ENERGIES_PRINT (CCENERGY)
CCENERGY — Do print MP2 and CCSD pair energies for RHF references?
- Type: boolean
- Default: false
- PARALLEL (SCF)
SCF (Expert) — Do run in parallel?
- Type: boolean
- Default: false
- PB_LAMBDA (DFTSAPT)
DFTSAPT — Lambda in Pauli Blockade
- Type: double
- Default: 1e5
- PCG_BETA_TYPE (OCC)
OCC — Type of PCG beta parameter (Fletcher-Reeves or Polak-Ribiere).
- Type: string
- Possible Values: FLETCHER_REEVES, POLAK_RIBIERE
- Default: FLETCHER_REEVES
- PCG_CONVERGENCE (OCC)
OCC — Convergence criterion for residual vector of preconditioned conjugate gradient method.
- Type: conv double
- Default: 1e-6
- PCG_MAXITER (OCC)
OCC — Maximum number of preconditioned conjugate gradient iterations.
- Type: integer
- Default: 30
- PERTURB_CBS (PSIMRCC)
PSIMRCC (Expert) — Do compute the perturbative corrections for basis set incompleteness?
- Type: boolean
- Default: false
- PERTURB_CBS_COUPLING (PSIMRCC)
PSIMRCC (Expert) — Do include the terms that couple different reference determinants in perturbative CBS correction computations?
- Type: boolean
- Default: true
- PERTURB_H (SCF)
SCF — Do perturb the Hamiltonian?
- Type: boolean
- Default: false
- PERTURB_MAGNITUDE (DETCI)
DETCI (Expert) — The magnitude of perturbation
 in
in 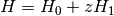
- Type: double
- Default: 1.0
- PERTURB_MAGNITUDE (SCF)
SCF — Size of the perturbation (applies only to dipole perturbations)
- Type: double
- Default: 0.0
- PERTURB_WITH (SCF)
SCF — The operator used to perturb the Hamiltonian, if requested
- Type: string
- Possible Values: DIPOLE_X, DIPOLE_Y, DIPOLE_Z, EMBPOT, SPHERE, DX
- Default: DIPOLE_X
- PHI_POINTS (SCF)
SCF — Number of azimuthal grid points for sphereical potential integration
- Type: integer
- Default: 360
- PITZER (TRANSQT)
TRANSQT — Do use Pitzer ordering?
- Type: boolean
- Default: false
- POINTS (FINDIF)
FINDIF — Number of points for finite-differences (3 or 5)
- Type: integer
- Default: 3
- POLE_MAXITER (ADC)
ADC — Maximum iteration number in pole searching
- Type: integer
- Default: 20
- PR (ADC)
ADC — Do use the partial renormalization scheme for the ground state wavefunction?
- Type: boolean
- Default: false
- PRECONDITIONER (DETCI)
DETCI — This specifies the type of preconditioner to use in the selected diagonalization method. The valid options are: DAVIDSON which approximates the Hamiltonian matrix by the diagonal elements; H0BLOCK_INV which uses an exact Hamiltonian of H0_BLOCKSIZE and explicitly inverts it; GEN_DAVIDSON which does a spectral decomposition of H0BLOCK; ITER_INV using an iterative approach to obtain the correction vector of H0BLOCK. The H0BLOCK_INV, GEN_DAVIDSON, and ITER_INV approaches are all formally equivalent but the ITER_INV is less computationally expensive. Default is DAVIDSON.
- Type: string
- Possible Values: LANCZOS, DAVIDSON, GEN_DAVIDSON, H0BLOCK, H0BLOCK_INV, ITER_INV, H0BLOCK_COUPLING, EVANGELISTI
- Default: DAVIDSON
- PRESORT_FILE (TRANSQT)
TRANSQT — SO-basis presort file
- Type: integer
- Default: PSIF_SO_PRESORT
- PRINT (CPHF)
CPHF — The amount of information printed to the output file
- Type: integer
- Default: 1
- PRINT (DFTSAPT)
DFTSAPT — The amount of information printed to the output file
- Type: integer
- Default: 1
- PRINT (GLOBALS)
GLOBALS — The amount of information to print to the output file. 1 prints basic information, and higher levels print more information. A value of 5 will print very large amounts of debugging information.
- Type: integer
- Default: 1
- PRINT (SAPT)
SAPT — The amount of information to print to the output file for the sapt module. For 0, only the header and final results are printed. For 1, (recommended for large calculations) some intermediate quantities are also printed.
- Type: integer
- Default: 1
- PRINT_BASIS (SCF)
SCF — Flag to print the basis set.
- Type: boolean
- Default: false
- PRINT_LVL (TRANSQT)
TRANSQT — The amount of information to print to the output file. 1 prints basic information, and higher levels print more information. A value of 5 will print very large amounts of debugging information.
- Type: integer
- Default: 1
- PRINT_MOS (SCF)
SCF — Flag to print the molecular orbitals.
- Type: boolean
- Default: false
- PRINT_MOS (TRANSQT)
TRANSQT — Do print MOs?
- Type: boolean
- Default: false
- PRINT_OE_INTEGRALS (TRANSQT)
TRANSQT — Do print one-electron integrals?
- Type: boolean
- Default: false
- PRINT_REORDER (TRANSQT)
TRANSQT — Do print reordered MOs?
- Type: boolean
- Default: false
- PRINT_SORTED_OE_INTS (TRANSQT)
TRANSQT — Do print sorted one-electron integrals?
- Type: boolean
- Default: false
- PRINT_SORTED_TE_INTS (TRANSQT)
TRANSQT — Do print sorted two-electron integrals (TEIs)?
- Type: boolean
- Default: false
- PRINT_TE_INTEGRALS (TRANSQT)
TRANSQT — Do print two-electron integrals?
- Type: boolean
- Default: false
- PRINT_TEI (TRANSQT2)
TRANSQT2 — Do print two-electron integrals (TEIs)?
- Type: boolean
- Default: false
- PROCESS_GRID (SCF)
SCF (Expert) — The dimension sizes of the processor grid
- Type: array
- Default: No Default
- PROP_ALL (CCDENSITY)
CCDENSITY — Compute non-relaxed properties for all excited states.
- Type: boolean
- Default: true
- PROP_ALL (CCLAMBDA)
CCLAMBDA — Compute unrelaxed properties for all excited states.
- Type: boolean
- Default: true
- PROP_ROOT (CCDENSITY)
CCDENSITY — Root number (within its irrep) for computing properties
- Type: integer
- Default: 1
- PROP_ROOT (CCEOM)
CCEOM — Root number (within its irrep) for computing properties. Defaults to highest root requested.
- Type: integer
- Default: 0
- PROP_ROOT (CCLAMBDA)
CCLAMBDA — Root number (within its irrep) for computing properties
- Type: integer
- Default: 1
- PROP_SYM (CCDENSITY)
CCDENSITY — The symmetry of states
- Type: integer
- Default: 1
- PROP_SYM (CCEOM)
CCEOM — Symmetry of the state to compute properties. Defaults to last irrep for which states are requested.
- Type: integer
- Default: 1
- PROP_SYM (CCLAMBDA)
CCLAMBDA — The symmetry of states
- Type: integer
- Default: 1
- PROPERTIES (GLOBALS)
GLOBALS — List of properties to compute
- Type: array
- Default: No Default
- PROPERTIES_ORIGIN (GLOBALS)
GLOBALS — Either a set of 3 coordinates, or a string (see manual) describing the origin about which one-electron properties are computed
- Type: array
- Default: No Default
- PROPERTY (CCENERGY)
CCENERGY — The response property desired. Acceptable values are POLARIZABILITY (default) for dipole-polarizabilities, ROTATION for specific rotations, ROA for Raman Optical Activity, and ALL for all of the above.
- Type: string
- Possible Values: POLARIZABILITY, ROTATION, MAGNETIZABILITY, ROA, ALL
- Default: POLARIZABILITY
- PROPERTY (CCRESPONSE)
CCRESPONSE — The response property desired. Acceptable values are POLARIZABILITY (default) for dipole-polarizabilities, ROTATION for specific rotations, ROA for Raman Optical Activity, and ALL for all of the above.
- Type: string
- Possible Values: POLARIZABILITY, ROTATION, ROA, ALL
- Default: POLARIZABILITY
- PROPERTY (CCSORT)
CCSORT — The response property desired. The unique acceptable values is POLARIZABILITY for dipole-polarizabilitie.
- Type: string
- Default: POLARIZABILITY
- PROPERTY (RESPONSE)
RESPONSE — Array that specifies the desired frequencies of the incident radiation field in CCLR calculations. If only one element is given, the units will be assumed to be atomic units. If more than one element is given, then the units must be specified as the final element of the array. Acceptable units are HZ, NM, EV, and AU.
- Type: string
- Possible Values: POLARIZABILITY, ROTATION, ROA, ALL
- Default: POLARIZABILITY
- PSIMRCC (TRANSQT)
TRANSQT — Do specific arrangements for PSIMRCC?
- Type: boolean
- Default: false
- PT_ENERGY (PSIMRCC)
PSIMRCC — The type of perturbation theory computation to perform
- Type: string
- Possible Values: SECOND_ORDER, SCS_SECOND_ORDER, PSEUDO_SECOND_ORDER, SCS_PSEUDO_SECOND_ORDER
- Default: SECOND_ORDER
- PUREAM (GLOBALS)
GLOBALS — Do use pure angular momentum basis functions? If not explicitly set, the default comes from the basis set.
- Type: boolean
- Default: true
- QC_COUPLING (DCFT)
DCFT — Controls whether to include the coupling terms in the DCFT electronic Hessian (for ALOGRITHM = QC only)
- Type: boolean
- Default: true
- QRHF (TRANSQT)
TRANSQT — Do form quasi RHF (QRHF) orbitals?
- Type: boolean
- Default: false
- R4S (DETCI)
DETCI (Expert) — Do restrict strings with
 in RAS IV? Useful to reduce the number of strings required if MIXED4=true, as in a split-virutal CISD[TQ] computation. If more than one electron is in RAS IV, then the holes in RAS I cannot exceed the number of particles in RAS III + RAS IV (i.e., EX_LEVEL), or else the string is discarded.
in RAS IV? Useful to reduce the number of strings required if MIXED4=true, as in a split-virutal CISD[TQ] computation. If more than one electron is in RAS IV, then the holes in RAS I cannot exceed the number of particles in RAS III + RAS IV (i.e., EX_LEVEL), or else the string is discarded.- Type: boolean
- Default: false
- R_CONVERGENCE (CCENERGY)
CCENERGY — Convergence criterion for wavefunction (change) in CC amplitude equations.
- Type: conv double
- Default: 1e-7
- R_CONVERGENCE (CCEOM)
CCEOM — Convergence criterion for norm of the residual vector in the Davidson algorithm for CC-EOM.
- Type: conv double
- Default: 1e-6
- R_CONVERGENCE (CCLAMBDA)
CCLAMBDA — Convergence criterion for wavefunction (change) in CC lambda-amplitude equations.
- Type: conv double
- Default: 1e-7
- R_CONVERGENCE (CCRESPONSE)
CCRESPONSE — Convergence criterion for wavefunction (change) in perturbed CC equations.
- Type: conv double
- Default: 1e-7
- R_CONVERGENCE (CIS)
CIS — Convergence criterion for CIS wavefunction.
- Type: conv double
- Default: 1e-7
- R_CONVERGENCE (DCFT)
DCFT — Convergence criterion for the RMS of the residual vector in the density cumulant updates, as well as the solution of the density cumulant and orbital response equations. In the orbital updates controls the RMS of the SCF error vector
- Type: conv double
- Default: 1e-10
- R_CONVERGENCE (DETCI)
DETCI — Convergence criterion for CI residual vector in the Davidson algorithm (RMS error). The default is 1e-4 for energies and 1e-7 for gradients.
- Type: conv double
- Default: 1e-4
- R_CONVERGENCE (FNOCC)
FNOCC — Convergence for the CC amplitudes. Note that convergence is met only when E_CONVERGENCE and R_CONVERGENCE are satisfied.
- Type: conv double
- Default: 1.0e-7
- R_CONVERGENCE (LMP2)
LMP2 — Convergence criterion for T2 amplitudes (RMS change).
- Type: conv double
- Default: 1e-5
- R_CONVERGENCE (OCC)
OCC — Convergence criterion for amplitudes (residuals).
- Type: conv double
- Default: 1e-5
- R_CONVERGENCE (PSIMRCC)
PSIMRCC — Convergence criterion for amplitudes (residuals).
- Type: conv double
- Default: 1e-9
- R_POINTS (SCF)
SCF — Number of radial grid points for sphereical potential integration
- Type: integer
- Default: 100
- RADIUS (SCF)
SCF — Radius (bohr) of a hard-sphere external potential
- Type: double
- Default: 10.0
- RAS1 (DETCI)
DETCI (Expert) — An array giving the number of orbitals per irrep for RAS1
- Type: array
- Default: No Default
- RAS1 (TRANSQT)
TRANSQT (Expert) — An array giving the number of orbitals per irrep for RAS1
- Type: array
- Default: No Default
- RAS2 (DETCI)
DETCI (Expert) — An array giving the number of orbitals per irrep for RAS2
- Type: array
- Default: No Default
- RAS2 (TRANSQT)
TRANSQT (Expert) — An array giving the number of orbitals per irrep for RAS2
- Type: array
- Default: No Default
- RAS3 (DETCI)
DETCI (Expert) — An array giving the number of orbitals per irrep for RAS3
- Type: array
- Default: No Default
- RAS3 (TRANSQT)
TRANSQT (Expert) — An array giving the number of orbitals per irrep for RAS3
- Type: array
- Default: No Default
- RAS34_MAX (DETCI)
DETCI — maximum number of electrons in RAS III + IV
- Type: integer
- Default: -1
- RAS3_MAX (DETCI)
DETCI — maximum number of electrons in RAS III
- Type: integer
- Default: -1
- RAS4 (DETCI)
DETCI (Expert) — An array giving the number of orbitals per irrep for RAS4
- Type: array
- Default: No Default
- RAS4 (TRANSQT)
TRANSQT (Expert) — An array giving the number of orbitals per irrep for RAS4
- Type: array
- Default: No Default
- RAS4_MAX (DETCI)
DETCI — maximum number of electrons in RAS IV
- Type: integer
- Default: -1
- REFERENCE (ADC)
ADC — Reference wavefunction type
- Type: string
- Possible Values: RHF
- Default: RHF
- REFERENCE (CCDENSITY)
CCDENSITY — Reference wavefunction type
- Type: string
- Default: RHF
- REFERENCE (CCENERGY)
CCENERGY — Reference wavefunction type
- Type: string
- Possible Values: RHF, ROHF, UHF
- Default: RHF
- REFERENCE (CCEOM)
CCEOM — Reference wavefunction type
- Type: string
- Possible Values: RHF, ROHF, UHF
- Default: RHF
- REFERENCE (CCRESPONSE)
CCRESPONSE — Reference wavefunction type
- Type: string
- Default: RHF
- REFERENCE (CCSORT)
CCSORT — Reference wavefunction type
- Type: string
- Default: RHF
- REFERENCE (CCTRIPLES)
CCTRIPLES — Reference wavefunction type
- Type: string
- Default: RHF
- REFERENCE (CIS)
CIS — Reference wavefunction type
- Type: string
- Possible Values: RHF, ROHF, UHF
- Default: RHF
- REFERENCE (DCFT)
DCFT — Reference wavefunction type
- Type: string
- Possible Values: UHF
- Default: UHF
- REFERENCE (DETCI)
DETCI — Reference wavefunction type
- Type: string
- Possible Values: RHF, ROHF
- Default: RHF
- REFERENCE (LMP2)
LMP2 — Reference wavefunction type
- Type: string
- Possible Values: RHF
- Default: RHF
- REFERENCE (MCSCF)
MCSCF — Reference wavefunction type
- Type: string
- Possible Values: RHF, ROHF, UHF, TWOCON, MCSCF, GENERAL
- Default: RHF
- REFERENCE (RESPONSE)
RESPONSE — Reference wavefunction type
- Type: string
- Default: RHF
- REFERENCE (SCF)
SCF — Reference wavefunction type
- Type: string
- Possible Values: RHF, ROHF, UHF, CUHF, RKS, UKS
- Default: RHF
- REFERENCE (STABILITY)
STABILITY — Reference wavefunction type
- Type: string
- Possible Values: RHF, UHF, ROHF
- Default: RHF
- REFERENCE (TRANSQT)
TRANSQT — Reference wavefunction type
- Type: string
- Default: RHF
- REFERENCE (TRANSQT2)
TRANSQT2 — Reference wavefunction type
- Type: string
- Default: RHF
- REFERENCE_SYM (DETCI)
DETCI (Expert) — Irrep for CI vectors; -1 = find automatically. This option allows the user to look for CI vectors of a different irrep than the reference. This probably only makes sense for Full CI, and it would probably not work with unit vector guesses. Numbering starts from zero for the totally-symmetric irrep.
- Type: integer
- Default: -1
- RELAX_GUESS_ORBITALS (DCFT)
DCFT (Expert) — Controls whether to relax the guess orbitals by taking the guess density cumulant and performing orbital update on the first macroiteration (for ALOGRITHM = TWOSTEP only)
- Type: boolean
- Default: false
- RELAX_TAU (DCFT)
DCFT (Expert) — Controls whether to relax tau during the cumulant updates or not
- Type: boolean
- Default: true
- REORDER (TRANSQT)
TRANSQT — Do reorder MOs?
- Type: boolean
- Default: false
- REPL_OTF (DETCI)
DETCI (Expert) — Do string replacements on the fly in DETCI? Can save a gigantic amount of memory (especially for truncated CI’s) but is somewhat flaky and hasn’t been tested for a while. It may work only works for certain classes of RAS calculations. The current code is very slow with this option turned on.
- Type: boolean
- Default: false
- RESPONSE_ALGORITHM (DCFT)
DCFT — The algorithm to use for the solution of the response equations for the analytic gradients and properties.
- Type: string
- Possible Values: TWOSTEP, SIMULTANEOUS
- Default: TWOSTEP
- RESTART (CCENERGY)
CCENERGY — Do restart the coupled-cluster iterations from old
and amplitudes? For geometry optimizations, Brueckner calculations, etc. the iterative solution of the CC amplitude equations may benefit considerably by reusing old vectors as initial guesses. Assuming that the MO phases remain the same between updates, the CC codes will, by default, re-use old vectors, unless the user sets RESTART = false.- Type: boolean
- Default: true
- RESTART (CCLAMBDA)
CCLAMBDA — Do restart the coupled-cluster iterations from old
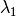 and
and 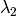 amplitudes?
amplitudes?- Type: boolean
- Default: false
- RESTART (CCRESPONSE)
CCRESPONSE — Do restart from on-disk amplitudes?
- Type: boolean
- Default: true
- RESTART (DETCI)
DETCI — Do restart a DETCI iteration that terminated prematurely? It assumes that the CI and sigma vectors are on disk; the number of vectors specified by RESTART_VECS (obsolete) is collapsed down to one vector per root.
- Type: boolean
- Default: false
- RESTART_EOM_CC3 (CCEOM)
CCEOM — Do restart from on-disk?
- Type: boolean
- Default: false
- RESTRICTED_DOCC (DETCI)
DETCI — An array giving the number of restricted doubly-occupied orbitals per irrep (not excited in CI wavefunctions, but orbitals can be optimized in MCSCF)
- Type: array
- Default: No Default
- RESTRICTED_DOCC (PSIMRCC)
PSIMRCC — The number of doubly occupied orbitals per irrep
- Type: array
- Default: No Default
- RESTRICTED_DOCC (TRANSQT)
TRANSQT — An array giving the number of restricted doubly-occupied orbitals per irrep (not excited in CI wavefunctions, but orbitals can be optimized in MCSCF)
- Type: array
- Default: No Default
- RESTRICTED_UOCC (DETCI)
DETCI — An array giving the number of restricted unoccupied orbitals per irrep (not occupied in CI wavefunctions, but orbitals can be optimized in MCSCF)
- Type: array
- Default: No Default
- RESTRICTED_UOCC (TRANSQT)
TRANSQT — An array giving the number of restricted unoccupied orbitals per irrep (not occupied in CI wavefunctions, but orbitals can be optimized in MCSCF)
- Type: array
- Default: No Default
- RFO_FOLLOW_ROOT (OPTKING)
OPTKING — Do follow the initial RFO vector after the first step?
- Type: boolean
- Default: false
- RFO_ROOT (OPTKING)
OPTKING — Root for RFO to follow, 0 being lowest (for a minimum)
- Type: integer
- Default: 0
- RHF_TRIPLETS (CCEOM)
CCEOM — Do form a triplet state from RHF reference?
- Type: boolean
- Default: false
- RMS_DISP_G_CONVERGENCE (OPTKING)
OPTKING — Convergence criterion for geometry optmization: rms displacement (internal coordinates, atomic units).
- Type: conv double
- Default: 1.2e-3
- RMS_FORCE_G_CONVERGENCE (OPTKING)
OPTKING — Convergence criterion for geometry optmization: rms force (internal coordinates, atomic units).
- Type: conv double
- Default: 3.0e-4
- RMS_MOGRAD_CONVERGENCE (OCC)
OCC — Convergence criterion for RMS orbital gradient. Default adjusts depending on E_CONVERGENCE.
- Type: conv double
- Default: 1e-6
- ROOTS_PER_IRREP (ADC)
ADC — The poles per irrep vector
- Type: array
- Default: No Default
- ROOTS_PER_IRREP (CCDENSITY)
CCDENSITY — The number of electronic states to computed, per irreducible representation
- Type: array
- Default: No Default
- ROOTS_PER_IRREP (CCEOM)
CCEOM — Number of excited states per irreducible representation for EOM-CC and CC-LR calculations. Irreps denote the final state symmetry, not the symmetry of the transition.
- Type: array
- Default: No Default
- ROOTS_PER_IRREP (CCLAMBDA)
CCLAMBDA — The number of electronic states to computed, per irreducible representation
- Type: array
- Default: No Default
- ROOTS_PER_IRREP (CIS)
CIS — The number of electronic states to computed, per irreducible representation
- Type: array
- Default: No Default
- ROTATE_MO_ANGLE (MCSCF)
MCSCF (Expert) — For orbital rotations after convergence, the angle (in degrees) by which to rotate.
- Type: double
- Default: 0.0
- ROTATE_MO_IRREP (MCSCF)
MCSCF (Expert) — For orbital rotations after convergence, irrep (1-based, Cotton order) of the orbitals to rotate.
- Type: integer
- Default: 1
- ROTATE_MO_P (MCSCF)
MCSCF (Expert) — For orbital rotations after convergence, number of the first orbital (1-based) to rotate.
- Type: integer
- Default: 1
- ROTATE_MO_Q (MCSCF)
MCSCF (Expert) — For orbital rotations after convergence, number of the second orbital (1-based) to rotate.
- Type: integer
- Default: 2
- ROTATION_SCHEME (STABILITY)
STABILITY — Method for following eigenvectors, either 0 by angles or 1 by antisymmetric matrix.
- Type: integer
- Default: 0
- RUN_CCSD (FNOCC)
FNOCC (Expert) — do ccsd rather than qcisd?
- Type: boolean
- Default: false
- RUN_CEPA (FNOCC)
FNOCC (Expert) — Is this a CEPA job? This parameter is used internally by the pythond driver. Changing its value won’t have any effect on the procedure.
- Type: boolean
- Default: false
- RUN_MP2 (FNOCC)
FNOCC (Expert) — do only evaluate mp2 energy?
- Type: boolean
- Default: false
- RUN_MP3 (FNOCC)
FNOCC (Expert) — do only evaluate mp3 energy?
- Type: boolean
- Default: false
- RUN_MP4 (FNOCC)
FNOCC (Expert) — do only evaluate mp4 energy?
- Type: boolean
- Default: false
- S (DETCI)
DETCI — The value of the spin quantum number
 is given by this option. The default is determined by the value of the multiplicity. This is used for two things: (1) determining the phase of the redundant half of the CI vector when the component is used (i.e., MS0 = TRUE), and (2) making sure the guess vector has the desired value of
is given by this option. The default is determined by the value of the multiplicity. This is used for two things: (1) determining the phase of the redundant half of the CI vector when the component is used (i.e., MS0 = TRUE), and (2) making sure the guess vector has the desired value of  (if S_SQUARED is TRUE and ICORE = 1).
(if S_SQUARED is TRUE and ICORE = 1).- Type: double
- Default: 0.0
- S_ORTHOGONALIZATION (SCF)
SCF — SO orthogonalization: symmetric or canonical?
- Type: string
- Possible Values: SYMMETRIC, CANONICAL
- Default: SYMMETRIC
- S_SQUARED (DETCI)
DETCI — Do calculate the value of
for each root? Only supported for ICORE = 1.- Type: boolean
- Default: false
- S_TOLERANCE (SCF)
SCF — Minimum S matrix eigenvalue to be used before compensating for linear dependencies.
- Type: conv double
- Default: 1e-7
- SAD_CHOL_TOLERANCE (SCF)
SCF (Expert) — SAD Guess Cholesky Cutoff (for eliminating redundancies).
- Type: conv double
- Default: 1e-7
- SAD_D_CONVERGENCE (SCF)
SCF — Convergence criterion for SCF density in SAD Guess.
- Type: conv double
- Default: 1e-5
- SAD_E_CONVERGENCE (SCF)
SCF — Convergence criterion for SCF energy in SAD Guess.
- Type: conv double
- Default: 1e-5
- SAD_F_MIX_START (SCF)
SCF (Expert) — SAD Guess F-mix Iteration Start
- Type: integer
- Default: 50
- SAD_MAXITER (SCF)
SCF (Expert) — Maximum number of SAD guess iterations
- Type: integer
- Default: 50
- SAD_PRINT (SCF)
SCF (Expert) — The amount of SAD information to print to the output
- Type: integer
- Default: 0
- SAPT (SCF)
SCF (Expert) — Are going to do SAPT? If so, what part?
- Type: string
- Possible Values: FALSE, 2-DIMER, 2-MONOMER_A, 2-MONOMER_B, 3-TRIMER, 3-DIMER_AB, 3-DIMER_BC, 3-DIMER_AC, 3-MONOMER_A, 3-MONOMER_B, 3-MONOMER_C
- Default: FALSE
- SAPT_LEVEL (SAPT)
SAPT — The level of theory for SAPT
- Type: string
- Possible Values: SAPT0, SAPT2, SAPT2+, SAPT2+3
- Default: SAPT0
- SAPT_MEM_CHECK (SAPT)
SAPT — Do force SAPT2 and higher to die if it thinks there isn’t enough memory? Turning this off is ill-advised.
- Type: boolean
- Default: true
- SAPT_MEM_FACTOR (DFTSAPT)
DFTSAPT — % of memory for DF-MP2 three-index buffers
- Type: double
- Default: 0.9
- SAPT_MEM_SAFETY (SAPT)
SAPT — Memory safety
- Type: double
- Default: 0.9
- SAPT_OS_SCALE (SAPT)
SAPT — The scale factor used for opposite-spin pairs in SCS computations. SS/OS decomposition performed for
and terms.- Type: double
- Default: 6.0/5.0
- SAPT_SS_SCALE (SAPT)
SAPT — The scale factor used for same-spin pairs in SCS computations. SS/OS decomposition performed for
and terms.- Type: double
- Default: 1.0/3.0
- SAVE_JK (SCF)
SCF — Keep JK object for later use?
- Type: boolean
- Default: false
- SCALE (STABILITY)
STABILITY — Scale factor (between 0 and 1) for orbital rotation step
- Type: double
- Default: 0.5
- SCF_MAXITER (DCFT)
DCFT — Maximum number of the orbital update micro-iterations per macro-iteration (for ALOGRITHM = TWOSTEP). Same keyword controls the maximum number of orbital response micro-iterations per macro-iteration for the solution of the response equations (for RESPONSE_ALOGRITHM = TWOSTEP)
- Type: integer
- Default: 50
- SCF_MEM_SAFETY_FACTOR (SCF)
SCF — Memory safety factor for allocating JK
- Type: double
- Default: 0.75
- SCF_TYPE (CPHF)
CPHF — SCF Type
- Type: string
- Possible Values: DIRECT, DF, PK, OUT_OF_CORE, PS
- Default: DIRECT
- SCF_TYPE (SCF)
SCF — What algorithm to use for the SCF computation. See Table SCF Convergence & Algorithm for default algorithm for different calculation types.
- Type: string
- Possible Values: DIRECT, DF, PK, OUT_OF_CORE
- Default: PK
- SCHMIDT_ADD_RESIDUAL_TOLERANCE (CCEOM)
CCEOM — Minimum absolute value above which a guess vector to a root is added to the Davidson algorithm in the EOM-CC iterative procedure.
- Type: conv double
- Default: 1e-3
- SCHWARZ_CUTOFF (CPHF)
CPHF — The schwarz cutoff value
- Type: double
- Default: 1.0e-12
- SCREEN_INTS (LMP2)
LMP2 — Do screen integrals?
- Type: boolean
- Default: false
- SCS (LMP2)
LMP2 — Do spin-component-scaled MP2 (SCS-MP2)?
- Type: boolean
- Default: false
- SCS_CCSD (CCENERGY)
CCENERGY — Do spin-component-scaled CCSD
- Type: boolean
- Default: false
- SCS_CCSD (FNOCC)
FNOCC — Do SCS-CCSD?
- Type: boolean
- Default: false
- SCS_CEPA (FNOCC)
FNOCC — Do SCS-CEPA? Note that the scaling factors will be identical to those for SCS-CCSD.
- Type: boolean
- Default: false
- SCS_MP2 (CCENERGY)
CCENERGY — Do spin-component-scaled MP2 (SCS-MP2)?
- Type: boolean
- Default: false
- SCS_MP2 (FNOCC)
FNOCC — Do SCS-MP2?
- Type: boolean
- Default: false
- SCS_N (LMP2)
LMP2 — Do SCS-MP2 with parameters optimized for nucleic acids?
- Type: boolean
- Default: false
- SCS_TYPE (OCC)
OCC — Type of the SCS method
- Type: string
- Possible Values: SCS, SCSN, SCSVDW, SCSMI
- Default: SCS
- SCSN_MP2 (CCENERGY)
CCENERGY — Do SCS-MP2 with parameters optimized for nucleic acids?
- Type: boolean
- Default: false
- SEKINO (CCLAMBDA)
CCLAMBDA — Do Sekino-Bartlett size-extensive model-III?
- Type: boolean
- Default: false
- SEKINO (CCRESPONSE)
CCRESPONSE — Do Sekino-Bartlett size-extensive model-III?
- Type: boolean
- Default: false
- SEM_MAXITER (ADC)
ADC — Maximum iteration number in simultaneous expansion method
- Type: integer
- Default: 30
- SEMICANONICAL (CCENERGY)
CCENERGY — Convert ROHF MOs to semicanonical MOs
- Type: boolean
- Default: true
- SEMICANONICAL (CCEOM)
CCEOM — Convert ROHF MOs to semicanonical MOs
- Type: boolean
- Default: true
- SEMICANONICAL (CCSORT)
CCSORT — Convert ROHF MOs to semicanonical MOs
- Type: boolean
- Default: true
- SEMICANONICAL (CCTRIPLES)
CCTRIPLES — Convert ROHF MOs to semicanonical MOs
- Type: boolean
- Default: true
- SEMICANONICAL (TRANSQT2)
TRANSQT2 — Convert ROHF MOs to semicanonical MOs
- Type: boolean
- Default: true
- SF_RESTRICT (DETCI)
DETCI (Expert) — Do eliminate determinants not valid for spin-complete spin-flip CI’s? [see J. S. Sears et al, J. Chem. Phys. 118, 9084-9094 (2003)]
- Type: boolean
- Default: false
- SIGMA_OVERLAP (DETCI)
DETCI (Expert) — Do print the sigma overlap matrix? Not generally useful.
- Type: boolean
- Default: false
- SINGLES_PRINT (CCEOM)
CCEOM — Do print information on the iterative solution to the single-excitation EOM-CC problem used as a guess to full EOM-CC?
- Type: boolean
- Default: false
- SMALL_CUTOFF (PSIMRCC)
PSIMRCC —
- Type: integer
- Default: 0
- SO_S_FILE (TRANSQT)
TRANSQT — SO basis overlap matrix file
- Type: integer
- Default: PSIF_OEI
- SO_T_FILE (TRANSQT)
TRANSQT — SO basis kinetic energy matrix file
- Type: integer
- Default: PSIF_OEI
- SO_TEI_FILE (TRANSQT)
TRANSQT — SO basis two-electron integrals file
- Type: integer
- Default: PSIF_SO_TEI
- SO_V_FILE (TRANSQT)
TRANSQT — SO basis potential energy matrix file
- Type: integer
- Default: PSIF_OEI
- SOCC (GLOBALS)
GLOBALS — An array containing the number of singly-occupied orbitals per irrep (in Cotton order). The value of DOCC should also be set.
- Type: array
- Default: No Default
- SOCC (MCSCF)
MCSCF — The number of singly occupied orbitals, per irrep
- Type: array
- Default: No Default
- SOLVER_CONVERGENCE (CPHF)
CPHF — Solver convergence threshold (max 2-norm).
- Type: conv double
- Default: 1.0e-6
- SOLVER_EXACT_DIAGONAL (CPHF)
CPHF — Solver exact diagonal or eigenvalue difference?
- Type: boolean
- Default: false
- SOLVER_MAX_SUBSPACE (CPHF)
CPHF — DL Solver maximum number of subspace vectors
- Type: integer
- Default: 6
- SOLVER_MAXITER (CPHF)
CPHF — Solver maximum iterations
- Type: integer
- Default: 100
- SOLVER_MIN_SUBSPACE (CPHF)
CPHF — DL Solver number of subspace vectors to collapse to
- Type: integer
- Default: 2
- SOLVER_N_GUESS (CPHF)
CPHF — DL Solver number of guesses
- Type: integer
- Default: 1
- SOLVER_N_ROOT (CPHF)
CPHF — DL Solver number of roots
- Type: integer
- Default: 1
- SOLVER_NORM (CPHF)
CPHF — DL Solver minimum corrector norm to add to subspace
- Type: double
- Default: 1.0e-6
- SOLVER_PRECONDITION (CPHF)
CPHF — Solver precondition type
- Type: string
- Possible Values: SUBSPACE, JACOBI, NONE
- Default: JACOBI
- SOLVER_PRECONDITION_MAXITER (CPHF)
CPHF — Solver precondtion max steps
- Type: integer
- Default: 1
- SOLVER_PRECONDITION_STEPS (CPHF)
CPHF — Solver precondition step type
- Type: string
- Possible Values: CONSTANT, TRIANGULAR
- Default: TRIANGULAR
- SOLVER_QUANTITY (CPHF)
CPHF — Solver residue or eigenvector delta
- Type: string
- Possible Values: EIGENVECTOR, RESIDUAL
- Default: RESIDUAL
- SOLVER_TYPE (CPHF)
CPHF — Solver type (for interchangeable solvers)
- Type: string
- Possible Values: DL, RAYLEIGH
- Default: DL
- SORTED_TEI_FILE (TRANSQT)
TRANSQT — MO-basis sorted two-electron integrals file
- Type: integer
- Default: PSIF_MO_TEI
- SOS_TYPE (OCC)
OCC — Type of the SOS method
- Type: string
- Possible Values: SOS, SOSPI
- Default: SOS
- SPINADAPT_ENERGIES (CCENERGY)
CCENERGY — Do print spin-adapted pair energies?
- Type: boolean
- Default: false
- SS_E_CONVERGENCE (CCEOM)
CCEOM — Convergence criterion for excitation energy (change) in the Davidson algorithm for the CIS guess to CC-EOM.
- Type: conv double
- Default: 1e-6
- SS_R_CONVERGENCE (CCEOM)
CCEOM — Convergence criterion for norm of the residual vector in the Davidson algorithm for the CIS guess to CC-EOM.
- Type: conv double
- Default: 1e-6
- SS_SKIP_DIAG (CCEOM)
CCEOM — Do skip diagonalization of Hbar SS block?
- Type: boolean
- Default: false
- SS_VECS_PER_ROOT (CCEOM)
CCEOM — SS vectors stored per root
- Type: integer
- Default: 5
- STABILITY_ADD_VECTORS (DCFT)
DCFT (Expert) — The number of vectors that can be added simultaneously into the subspace for Davidson’s diagonalization in stability check
- Type: integer
- Default: 20
- STABILITY_ANALYSIS (SCF)
SCF — Whether to perform stability analysis after convergence. NONE prevents analysis being performed. CHECK will print out the analysis of the wavefunction stability at the end of the computation. FOLLOW will perform the analysis and, if a totally symmetric instability is found, will attemp to follow the eigenvector and re-run the computations to find a stable solution.
- Type: string
- Possible Values: NONE, CHECK, FOLLOW
- Default: NONE
- STABILITY_AUGMENT_SPACE_TOL (DCFT)
DCFT (Expert) — The value of the rms of the residual in Schmidt orthogonalization which is used as a threshold for augmenting the vector subspace in stability check
- Type: conv double
- Default: 0.1
- STABILITY_CHECK (DCFT)
DCFT (Expert) — Performs stability analysis of the DCFT energy
- Type: boolean
- Default: false
- STABILITY_CONVERGENCE (DCFT)
DCFT (Expert) — Controls the convergence of the Davidson’s diagonalization in stability check
- Type: conv double
- Default: 1e-4
- STABILITY_MAX_SPACE_SIZE (DCFT)
DCFT (Expert) — The maximum size of the subspace for the stability check. The program will terminate if this parameter is exceeded and the convergence (STABILITY_CONVERGENCE) is not satisfied
- Type: integer
- Default: 200
- STABILITY_N_EIGENVALUES (DCFT)
DCFT (Expert) — The number of Hessian eigenvalues computed during the stability check
- Type: integer
- Default: 3
- STABILITY_N_GUESS_VECTORS (DCFT)
DCFT (Expert) — The number of guess vectors used for Davidson’s diagonalization in stability check
- Type: integer
- Default: 20
- STEP_TYPE (OPTKING)
OPTKING — Geometry optimization step type, either Newton-Raphson or Rational Function Optimization
- Type: string
- Possible Values: RFO, NR, SD, LINESEARCH_STATIC
- Default: RFO
- T (THERMO)
THERMO — Temperature in Kelvin for thermodynamic analysis.
- Type: double
- Default: 298.15
- T2_COUPLED (CCENERGY)
CCENERGY —
- Type: boolean
- Default: false
- T3_WS_INCORE (CCENERGY)
CCENERGY — Do build W intermediates required for cc3 in core memory?
- Type: boolean
- Default: false
- T3_WS_INCORE (CCEOM)
CCEOM — Do build W intermediates required for eom_cc3 in core memory?
- Type: boolean
- Default: false
- T_AMPS (CCHBAR)
CCHBAR — Do compute the Tamplitude equation matrix elements?
- Type: boolean
- Default: false
- TDHF_MEM_SAFETY_FACTOR (CPHF)
CPHF — Memory safety factor for allocating JK
- Type: double
- Default: 0.75
- TDM (DETCI)
DETCI — Do compute the transition density? Note: only transition densities between roots of the same symmetry will be evaluated. DETCI does not compute states of different irreps within the same computation; to do this, lower the symmetry of the computation.
- Type: boolean
- Default: false
- TDM_PRINT (DETCI)
DETCI — Do print the transition density?
- Type: boolean
- Default: false
- TDM_WRITE (DETCI)
DETCI — Do write the transition density?
- Type: boolean
- Default: false
- TEST_B (OPTKING)
OPTKING — Do test B matrix?
- Type: boolean
- Default: false
- TEST_DERIVATIVE_B (OPTKING)
OPTKING — Do test derivative B matrix?
- Type: boolean
- Default: false
- THETA_POINTS (SCF)
SCF — Number of colatitude grid points for sphereical potential integration
- Type: integer
- Default: 360
- THICKNESS (SCF)
SCF — Thickness (bohr) of a hard-sphere external potential
- Type: double
- Default: 20.0
- TIKHONOW_MAX (PSIMRCC)
PSIMRCC — The cycle after which Tikhonow regularization is stopped. Set to zero to allow regularization in all iterations
- Type: integer
- Default: 5
- TIKHONOW_OMEGA (DCFT)
DCFT (Expert) — The shift applied to the denominator in the density cumulant update iterations
- Type: double
- Default: 0.0
- TIKHONOW_OMEGA (PSIMRCC)
PSIMRCC — The shift to apply to the denominators, {it c.f.} Taube and Bartlett, JCP, 130, 144112 (2009)
- Type: double
- Default: 0.0
- TIKHONOW_TRIPLES (PSIMRCC)
PSIMRCC (Expert) — Do use Tikhonow regularization in (T) computations?
- Type: boolean
- Default: false
- TILE_SZ (SCF)
SCF (Expert) — The tile size for the distributed matrices
- Type: integer
- Default: 512
- TPDM (DCFT)
DCFT (Expert) — Controls whether to compute unrelaxed two-particle density matrix at the end of the energy computation
- Type: boolean
- Default: false
- TPDM (DETCI)
DETCI — Do compute two-particle density matrix if not otherwise required?
- Type: boolean
- Default: false
- TPDM_ABCD_TYPE (OCC)
OCC — How to take care of the TPDM VVVV-block. The COMPUTE option means it will be computed via an IC/OOC algoritm. The DIRECT option (default) means it will not be computed and stored, instead its contribution will be directly added to Generalized-Fock Matrix.
- Type: string
- Possible Values: DIRECT, COMPUTE
- Default: DIRECT
- TPDM_ADD_REF (TRANSQT)
TRANSQT — Do add reference contribution to TPDM?
- Type: boolean
- Default: false
- TPDM_FILE (TRANSQT)
TRANSQT — MO-basis two-particle density matrix file
- Type: integer
- Default: PSIF_MO_TPDM
- TPDM_PRINT (DETCI)
DETCI — Do print the two-particle density matrix? (Warning: large tensor)
- Type: boolean
- Default: false
- TRIPLES_ALGORITHM (PSIMRCC)
PSIMRCC — The type of algorithm to use for (T) computations
- Type: string
- Possible Values: SPIN_ADAPTED, RESTRICTED, UNRESTRICTED
- Default: RESTRICTED
- TRIPLES_DIIS (PSIMRCC)
PSIMRCC — Do use DIIS extrapolation to accelerate convergence for iterative triples excitations?
- Type: boolean
- Default: false
- TRIPLES_LOW_MEMORY (FNOCC)
FNOCC — Do use low memory option for triples contribution? Note that this option is enabled automatically if the memory requirements of the conventional algorithm would exceed the available resources
- Type: boolean
- Default: false
- TURN_ON_ACTV (MCSCF)
MCSCF —
- Type: integer
- Default: 0
- UNITS (GLOBALS)
GLOBALS — Units used in geometry specification
- Type: string
- Possible Values: BOHR, AU, A.U., ANGSTROMS, ANG, ANGSTROM
- Default: ANGSTROMS
- UPDATE (DETCI)
DETCI — The update or correction vector formula, either DAVIDSON (default) or OLSEN.
- Type: string
- Possible Values: DAVIDSON, OLSEN
- Default: DAVIDSON
- USE_SPIN_SYM (PSIMRCC)
PSIMRCC — Do use symmetry to map equivalent determinants onto each other, for efficiency?
- Type: boolean
- Default: true
- USE_SPIN_SYMMETRY (PSIMRCC)
PSIMRCC (Expert) — Whether to use spin symmetry to map equivalent configurations onto each other, for efficiency
- Type: boolean
- Default: true
- VAL_EX_LEVEL (DETCI)
DETCI — In a RAS CI, this is the additional excitation level for allowing electrons out of RAS I into RAS II. The maximum number of holes in RAS I is therefore EX_LEVEL + VAL_EX_LEVEL.
- Type: integer
- Default: 0
- VECS_CC3 (CCEOM)
CCEOM — Vectors stored in CC3 computations
- Type: integer
- Default: 10
- VECS_PER_ROOT (CCEOM)
CCEOM — Vectors stored per root
- Type: integer
- Default: 12
- VECS_WRITE (DETCI)
DETCI — Do store converged vector(s) at the end of the run? The vector(s) is(are) stored in a transparent format such that other programs can use it easily. The format is specified in psi4/src/lib/libqt/slaterdset.h .
- Type: boolean
- Default: false
- WABEI_LOWDISK (CCHBAR)
CCHBAR — Do use the minimal-disk algorithm for Wabei? It’s VERY slow!
- Type: boolean
- Default: false
- WFN (CCDENSITY)
CCDENSITY (Expert) — Wavefunction type
- Type: string
- Default: SCF
- WFN (CCENERGY)
CCENERGY (Expert) — Wavefunction type
- Type: string
- Possible Values: CCSD, CCSD_T, EOM_CCSD, LEOM_CCSD, BCCD, BCCD_T, CC2, CC3, EOM_CC2, EOM_CC3, CCSD_MVD
- Default: NONE
- WFN (CCEOM)
CCEOM (Expert) — Wavefunction type
- Type: string
- Possible Values: EOM_CCSD, EOM_CC2, EOM_CC3
- Default: EOM_CCSD
- WFN (CCHBAR)
CCHBAR (Expert) — Wavefunction type
- Type: string
- Default: SCF
- WFN (CCLAMBDA)
CCLAMBDA (Expert) — Wavefunction type
- Type: string
- Default: SCF
- WFN (CCRESPONSE)
CCRESPONSE (Expert) — Wavefunction type
- Type: string
- Default: SCF
- WFN (CCSORT)
CCSORT (Expert) — Wavefunction type
- Type: string
- Default: No Default
- WFN (CCTRIPLES)
CCTRIPLES (Expert) — Wavefunction type
- Type: string
- Default: SCF
- WFN (CIS)
CIS (Expert) — Wavefunction type
- Type: string
- Possible Values: CCSD, CCSD_T, EOM_CCSD, CIS
- Default: CIS
- WFN (CLAG)
CLAG (Expert) — Wavefunction type
- Type: string
- Default: NONE
- WFN (DETCI)
DETCI (Expert) — Wavefunction type. This should be set automatically from the calling Psithon function.
- Type: string
- Possible Values: DETCI, CI, ZAPTN, DETCAS, CASSCF, RASSCF
- Default: DETCI
- WFN (GLOBALS)
GLOBALS (Expert) — Wavefunction type
- Type: string
- Default: SCF
- WFN (LMP2)
LMP2 (Expert) — Wavefunction type
- Type: string
- Default: LMP2
- WFN (SCF)
SCF (Expert) — Wavefunction type
- Type: string
- Possible Values: SCF
- Default: SCF
- WFN (TRANSQT)
TRANSQT (Expert) — Wavefunction type
- Type: string
- Default: CCSD
- WFN (TRANSQT2)
TRANSQT2 (Expert) — Wavefunction type
- Type: string
- Default: No Default
- WFN_SYM (MCSCF)
MCSCF — The symmetry of the SCF wavefunction.
- Type: string
- Possible Values: A, AG, AU, AP, APP, A1, A2, B, BG, BU, B1, B2, B3, B1G, B2G, B3G, B1U, B2U, B3U, 0, 1, 2, 3, 4, 5, 6, 7, 8
- Default: 1
- WFN_SYM (PSIMRCC)
PSIMRCC — The symmetry of the target wavefunction, specified either by Schönflies symbol, or irrep number (in Cotton ordering)
- Type: string
- Possible Values: A, AG, AU, AP, APP, A1, A2, B, BG, BU, B1, B2, B3, B1G, B2G, B3G, B1U, B2U, B3U, 0, 1, 2, 3, 4, 5, 6, 7, 8
- Default: 1
- WFN_TYPE (OCC)
OCC — Type of the wavefunction.
- Type: string
- Possible Values: OMP2, OMP3, OCEPA, OMP2.5
- Default: OMP2
- WRITER_FILE_LABEL (GLOBALS)
GLOBALS — Base filename for text files written by PSI, such as the MOLDEN output file, the Hessian file, the internal coordinate file, etc. Use the add_str_i function to make this string case sensitive.
- Type: string
- Default: No Default
- XI (CCDENSITY)
CCDENSITY — Do compute Xi?
- Type: boolean
- Default: false
- XI_CONNECT (CCDENSITY)
CCDENSITY (Expert) — Do require
 and
and  to be connected?
to be connected?- Type: boolean
- Default: false
- ZERO_INTERNAL_AMPS (PSIMRCC)
PSIMRCC — Do zero the internal amplitudes, i.e., those that map reference determinants onto each other?
- Type: boolean
- Default: true
- ZETA (CCDENSITY)
CCDENSITY — Do use zeta?
- Type: boolean
- Default: false
- ZETA (CCLAMBDA)
CCLAMBDA — Do use zeta?
- Type: boolean
- Default: false