
Python Driver¶
basislist¶
Module to define a class BasisFamily that associates fitting basis sets to an orbital basis and to provide functions to query appropriate fitting bases for any orbital basis distributed with Psi4.
- class BasisFamily(ornate, orbital=None, jk=None, ri=None, dual=None)[source]¶
Class to associate with an orbital basis name ornate the gbs file names in which the orbital basis orbital (usually the coded form of ornate) and jkfit, rifit, and dualfit auxiliary bases can be found.
- add_dualfit(fit)[source]¶
Function to add basis fit as associated helper basis member dualfit to a BasisFamily object.
- add_jkfit(fit)[source]¶
Function to add basis fit as associated fitting basis member jkfit to a BasisFamily object.
- corresponding_dualfit(name)[source]¶
Function to return an appropriate DUAL helper basis for the orbital basis name in coded or ornate form. None is returned if no fitting basis is defined or if the orbital basis is not found.
- corresponding_jkfit(name)[source]¶
Function to return an appropriate JK fitting basis for the orbital basis name in coded or ornate form. None is returned if no fitting basis is defined or if the orbital basis is not found.
- corresponding_rifit(name)[source]¶
Function to return an appropriate RI fitting basis for the orbital basis name in coded or ornate form. None is returned if no fitting basis is defined or if the orbital basis is not found.
- load_basis_families()[source]¶
Function to load into the array basisfamily_list BasisFamily objects for all Psi4’s standard installed bases.
input¶
- check_parentheses_and_brackets(input_string, exit_on_error)[source]¶
Function to check that all parenthesis and brackets in input_string are paired. On that condition, exit_on_error =1, otherwise 0.
- parse_multiline_array(input_list)[source]¶
Function to squash multiline arrays into a single line until all parentheses and brackets are fully paired.
- process_input(raw_input, print_level=1)[source]¶
Function to preprocess raw input, the text of the input file, then parse it, validate it for format, and convert it into legitimate Python. raw_input is printed to the output file unless print_level =0. Does a series of regular expression filters, where the matching portion of the input is replaced by the output of the corresponding function (in this module) call. Returns a string concatenating module import lines, a copy of the user’s .psi4rc files, a setting of the scratch directory, a dummy molecule, and the processed raw_input.
- process_multiline_arrays(inputfile)[source]¶
Function to find array inputs that are spread across multiple lines and squash them into a single line.
- process_option(spaces, module, key, value, line)[source]¶
Function to process a line with set or in a set block into global/local domain and keyword/value.
- process_print_command(matchobj)[source]¶
Function to process match of print and transform it to PsiMod.print_out().
- process_set_command(matchobj)[source]¶
Function to process match of all individual set (module_list) key {[value_list] or $value or value}.
pubchem¶
psiexceptions¶
Module with non-generic exceptions classes.
frac¶
molutil¶
Module with utility functions that act on molecule objects.
- BFS(self)¶
Perform a breadth-first search (BFS) on the real atoms in molecule, returning an array of atom indices of fragments. Relies upon van der Waals radii and so faulty for close (esp. hydrogen-bonded) fragments. Original code from Michael S. Marshall.
- dynamic_variable_bind(cls)[source]¶
Function to dynamically add extra members to the PsiMod.Molecule class.
- extract_cluster_indexing(mol, cluster_size=0)[source]¶
Function to returns a LIST of all subclusters of the molecule mol of real size cluster_size. If cluster_size = 0, returns all possible combinations of cluster size.
- extract_clusters(mol, ghost=True, cluster_size=0)[source]¶
Function to return all subclusters of the molecule mol of real size cluster_size and all other atoms ghosted if ghost equals true, all other atoms discarded if ghost is false. If cluster_size = 0, returns all possible combinations of cluster size.
proc¶
- run_adc(name, **kwargs)[source]¶
Function encoding sequence of PSI module calls for an algebraic diagrammatic construction calculation.
Caution
Get rid of active molecule lines- should be handled in energy.
- run_bccd(name, **kwargs)[source]¶
Function encoding sequence of PSI module calls for a Brueckner CCD calculation.
- run_bccd_t(name, **kwargs)[source]¶
Function encoding sequence of PSI module calls for a Brueckner CCD(T) calculation.
- run_cc_gradient(name, **kwargs)[source]¶
Function encoding sequence of PSI module calls for a CCSD and CCSD(T) gradient calculation.
- run_cc_property(name, **kwargs)[source]¶
Function encoding sequence of PSI module calls for all CC property calculations.
- run_ccenergy(name, **kwargs)[source]¶
Function encoding sequence of PSI module calls for a CCSD, CC2, and CC3 calculation.
- run_cepa(name, **kwargs)[source]¶
Function encoding sequence of PSI module calls for a cepa-like calculation.
>>> energy('cepa(1)')
- run_dcft(name, **kwargs)[source]¶
Function encoding sequence of PSI module calls for a density cumulant functional theory calculation.
- run_dcft_gradient(name, **kwargs)[source]¶
Function encoding sequence of PSI module calls for DCFT gradient calculation.
- run_detci(name, **kwargs)[source]¶
Function encoding sequence of PSI module calls for a configuration interaction calculation, namely FCI, CIn, MPn, and ZAPTn.
- run_dfmp2(name, **kwargs)[source]¶
Function encoding sequence of PSI module calls for a density-fitted MP2 calculation.
- run_dfmp2_gradient(name, **kwargs)[source]¶
Function encoding sequence of PSI module calls for a DFMP2 gradient calculation.
- run_dft(name, **kwargs)[source]¶
Function encoding sequence of PSI module calls for a density-functional-theory calculation.
- run_dft_gradient(name, **kwargs)[source]¶
Function encoding sequence of PSI module calls for a density-functional-theory gradient calculation.
- run_eom_cc(name, **kwargs)[source]¶
Function encoding sequence of PSI module calls for an EOM-CC calculation, namely EOM-CC2, EOM-CCSD, and EOM-CC3.
- run_eom_cc_gradient(name, **kwargs)[source]¶
Function encoding sequence of PSI module calls for an EOM-CCSD gradient calculation.
- run_libfock(name, **kwargs)[source]¶
Function encoding sequence of PSI module calls for a calculation through libfock, namely RCPHF, RCIS, RTDHF, RTDA, and RTDDFT.
- run_mcscf(name, **kwargs)[source]¶
Function encoding sequence of PSI module calls for a multiconfigurational self-consistent-field calculation.
- run_mp2(name, **kwargs)[source]¶
Function encoding sequence of PSI module calls for a MP2 calculation.
- run_mp2_gradient(name, **kwargs)[source]¶
Function encoding sequence of PSI module calls for a MP2 gradient calculation.
- run_mp2_select(name, **kwargs)¶
Function selecting the algorithm for a MP2 energy call and directing toward the MP2 or the DFMP2 modules.
- run_mp2_select_gradient(name, **kwargs)¶
Function selecting the algorithm for a MP2 gradient call and directing toward the MP2 or the DFMP2 modules.
- run_mp2c(name, **kwargs)[source]¶
Function encoding sequence of PSI module calls for a coupled MP2 calculation.
- run_mrcc(name, **kwargs)[source]¶
Function that prepares environment and input files for a calculation calling Kallay’s MRCC code.
- run_omp2(name, **kwargs)[source]¶
Function encoding sequence of PSI module calls for an orbital-optimized MP2 computation
- run_omp2_gradient(name, **kwargs)¶
Function encoding sequence of PSI module calls for OMP2 gradient calculation.
- run_omp3(name, **kwargs)[source]¶
Function encoding sequence of PSI module calls for an orbital-optimized MP3 computation
- run_psimrcc(name, **kwargs)[source]¶
Function encoding sequence of PSI module calls for a PSIMRCC computation using a reference from the MCSCF module
- run_psimrcc_scf(name, **kwargs)[source]¶
Function encoding sequence of PSI module calls for a PSIMRCC computation using a reference from the SCF module
- run_sapt(name, **kwargs)[source]¶
Function encoding sequence of PSI module calls for a SAPT calculation of any level.
- run_sapt_ct(name, **kwargs)[source]¶
Function encoding sequence of PSI module calls for a charge-transfer SAPT calcuation of any level.
- run_scf(name, **kwargs)[source]¶
Function encoding sequence of PSI module calls for a self-consistent-field theory (HF & DFT) calculation.
- run_scf_gradient(name, **kwargs)[source]¶
Function encoding sequence of PSI module calls for a SCF gradient calculation.
- run_scf_property(name, **kwargs)[source]¶
Function encoding sequence of PSI module calls for SCF calculations. This is a simple alias to run_scf() since SCF properties all handled through oeprop.
- run_scs_omp2(name, **kwargs)¶
Function encoding sequence of PSI module calls for a spin-component scaled OMP2 computation
- run_scs_omp3(name, **kwargs)¶
Function encoding sequence of PSI module calls for a spin-component scaled OMP3 computation
- run_sos_omp2(name, **kwargs)¶
Function encoding sequence of PSI module calls for a spin-opposite scaled OMP2 computation
- run_sos_omp3(name, **kwargs)¶
Function encoding sequence of PSI module calls for a spin-opposite scaled OMP3 computation
dashparam¶
Module to hold and distribute the -D dispersion correction parameters.
- dash_server(func, dashlvl, mode='psi4')[source]¶
Function to serve up dispersion correction parameters in whatever form needed. When mode is ‘dftd3’, returns a string suitable for writing to ./dftd3_parameters to calculuate the correction at dashlvl with the default parameters for functional func. When mode is ‘psi4’, returns a tuple of arguments suitable for building a Dispersion object with dashlvl parameters for functional func.
There are four computational dashlvl choices. ‘d2p4’ calls the -D2 correction within psi4 (hence, faked for mode=’dftd3’). The other three, ‘d2gr’, ‘d3zero’, and ‘d3bj’ call the three dftd3 modes of operation (corresponding to -old, -zero, -bj). Additionally, there are three aliased dashlvl choices since the aliases in dash_alias above are imposed.
procutil¶
Module with utility functions used by several Python functions.
- all_casings(input_string)[source]¶
Function to return a generator of all lettercase permutations of input_string.
- drop_duplicates(seq)[source]¶
Function that given an array seq, returns an array without any duplicate entries. There is no guarantee of which duplicate entry is dropped.
- format_kwargs_for_input(filename, lmode=1, **kwargs)[source]¶
Function to pickle to file filename the options dictionary kwargs. Mode lmode =2 pickles appropriate settings for reap mode. Used to capture Python options information for distributed (sow/reap) input files.
- format_molecule_for_input(mol)[source]¶
Function to return a string of the output of input.process_input() applied to the XYZ format of molecule, passed as either fragmented geometry string mol or molecule instance mol. Used to capture molecule information from database modules and for distributed (sow/reap) input files. For the reverse, see molutil.geometry().
- format_options_for_input()[source]¶
Function to return a string of commands to replicate the current state of user-modified options. Used to capture C++ options information for distributed (sow/reap) input files.
Caution
Some features are not yet implemented. Buy a developer a coffee.
- Does not cover local (as opposed to global) options.
- Does not work with array-type options.
- get_psifile(fileno, pidspace='18820')[source]¶
Function to return the full path and filename for psi file fileno (e.g., psi.32) in current namespace pidspace.
- getattr_ignorecase(module, attr)[source]¶
Function to extract attribute attr from module if attr is available in any possible lettercase permutation. Returns attribute if available, None if not.
driver¶
- gradient(name, **kwargs)[source]¶
Function complementary to optimize(). Carries out one gradient pass, deciding analytic or finite difference.
- hessian(name, **kwargs)[source]¶
Function to compute force constants. Presently identical to frequency().
optproc¶
Module to provide mechanism to store and restore option states in driver.
- class OptionState(option, module=None)[source]¶
Class to store the state of a single option. If module given, the option value and has_changed value is stored for global, local to module, and used by module scopes; otherwise (used for BASIS keywords), only global scope is stored. Class can store, print, and restore option values.
>>> OptionState('SCF_TYPE', 'SCF') >>> print(OptionState('DF_BASIS_MP2'))
- class OptionsState(*largs)[source]¶
Class to contain multiple OptionsState() objects. Used in python driver functions to collect several options before altering them, then restoring before function return.
>>> optstash = OptionsState( ['SCF', 'DFT_FUNCTIONAL'], ['DF_BASIS_SCF'], ['SCF', 'SCF_TYPE'], ['SCF', 'REFERENCE']) >>> print(optstash) >>> optstash.restore()
physconst¶
# Do not modify this file! It is auto-generated by the document_options_and_tests
# script, from psi4topdir/include/physconst.h
psi_h = 6.62606896E-34 # The Planck constant (Js)
psi_c = 2.99792458E8 # Speed of light (ms$^{-1}$)
psi_kb = 1.3806504E-23 # The Boltzmann constant (JK$^{-1}$)
psi_R = 8.314472 # Universal gas constant (JK$^{-1}$mol$^{-1}$)
psi_bohr2angstroms = 0.52917720859 # Bohr to Angstroms conversion factor
psi_bohr2m = 0.52917720859E-10 # Bohr to meters conversion factor
psi_bohr2cm = 0.52917720859E-8 # Bohr to centimeters conversion factor
psi_amu2g = 1.660538782E-24 # Atomic mass units to grams conversion factor
psi_amu2kg = 1.660538782E-27 # Atomic mass units to kg conversion factor
psi_au2amu = 5.485799097E-4 # Atomic units (m$@@e$) to atomic mass units conversion factor
psi_hartree2J = 4.359744E-18 # Hartree to joule conversion factor
psi_hartree2aJ = 4.359744 # Hartree to attojoule (10$^{-18}$J) conversion factor
psi_cal2J = 4.184 # Calorie to joule conversion factor
psi_dipmom_au2si = 8.47835281E-30 # Atomic units to SI units (Cm) conversion factor for dipoles
psi_dipmom_au2debye = 2.54174623 # Atomic units to Debye conversion factor for dipoles
psi_dipmom_debye2si = 3.335640952E-30 # Debye to SI units (Cm) conversion factor for dipoles
psi_c_au = 137.035999679 # Speed of light in atomic units
psi_hartree2ev = 27.21138 # Hartree to eV conversion factor
psi_hartree2wavenumbers = 219474.6 # Hartree to cm$^{-1}$ conversion factor
psi_hartree2kcalmol = 627.5095 # Hartree to kcal mol$^{-1}$ conversion factor
psi_hartree2MHz = 6.579684E9 # Hartree to MHz conversion factor
psi_kcalmol2wavenumbers = 349.7551 # kcal mol$^{-1}$ to cm$^{-1}$ conversion factor
psi_e0 = 8.854187817E-12 # Vacuum permittivity (Fm$^{-1}$)
psi_na = 6.02214179E23 # Avagadro's number
psi_me = 9.10938215E-31 # Electron rest mass (in kg)
wrappers¶
Module with functions that call the four main driver functions: driver.energy, driver.optimize, driver.response, and driver.frequency.
- auto_fragments(name, **kwargs)[source]¶
Detects fragments if the user does not supply them. Currently only used for the WebMO implementation of SAPT
usage: auto_fragments(‘’)
- call_function_in_1st_argument(funcarg, **largs)[source]¶
Function to make primary function call to energy(), opt(), etc. with options dictionary largs. Useful when funcarg to call is stored in variable.
- reconstitute_bracketed_basis(needarray)[source]¶
Function to reform a bracketed basis set string from a sequential series of basis sets (e.g, form ‘cc-pv[q5]z’ from array [cc-pvqz, cc-pv5z]). The basis set array is extracted from the f_basis field of a NEED dictionary in complete_basis_set(). Result is used to print a nicely formatted basis set string in the results table.
- split_menial(menial)[source]¶
Function used by complete_basis_set() to separate menial ‘scftot’ into [scf, tot] and ‘mp2corl’ into [mp2, corl].
- tblhead(tbl_maxrgt, tbl_delimit, ttype)[source]¶
Function that prints the header for the changable-width results tables in db(). tbl_maxrgt is the number of reagent columns the table must plan for. tbl_delimit is a string of dashes of the correct length to set off the table. ttype is 1 for tables comparing the computed values to the reference or 2 for simple tabulation and sum of the computed values.
- validate_bracketed_basis(basisstring)[source]¶
Function to transform and validate basis sets for cbs(). A basis set with no paired square brackets is passed through with zeta level 0 (e.g., ‘6-31+G(d,p)’ is returned as [6-31+G(d,p)] and [0]). A basis set with square brackets is checked for sensible sequence and Dunning-ness and returned as separate basis sets (e.g., ‘cc-pV[Q5]Z’ is returned as [cc-pVQZ, cc-pV5Z] and [4, 5]). Note that this function has no communication with the basis set library to check if the basis actually exists. Used by complete_basis_set().
- validate_scheme_args(functionname, **largs)[source]¶
Function called by each extrapolation scheme in complete_basis_set(). Checks that all the input arguments are present and suitable so that the scheme function can focus on defining the extrapolation.
qmmm¶
Module with classes to integrate MM charges into a QM calculation.
basislistother¶
Module with commands building BasisFamily objects for Pople and other non-Dunning orbital basis sets. Some plausible fitting basis sets are supplied as defaults.
diatomic¶
functional¶
Module to provide lightweight definitions of functionals and SuperFunctionals
- build_b2plypd2gr_superfunctional(name, npoints, deriv)¶
- build_b2plypd2p4_superfunctional(name, npoints, deriv)¶
- build_b2plypd3bj_superfunctional(name, npoints, deriv)¶
- build_b2plypd3zero_superfunctional(name, npoints, deriv)¶
- build_b3lyp5d2gr_superfunctional(name, npoints, deriv)¶
- build_b3lyp5d2p4_superfunctional(name, npoints, deriv)¶
- build_b3lyp5d3bj_superfunctional(name, npoints, deriv)¶
- build_b3lyp5d3zero_superfunctional(name, npoints, deriv)¶
- build_b3lypd1_superfunctional(name, npoints, deriv)¶
- build_b3lypd2gr_superfunctional(name, npoints, deriv)¶
- build_b3lypd2p4_superfunctional(name, npoints, deriv)¶
- build_b3lypd3bj_superfunctional(name, npoints, deriv)¶
- build_b3lypd3zero_superfunctional(name, npoints, deriv)¶
- build_b97d2gr_superfunctional(name, npoints, deriv)¶
- build_b97d2p4_superfunctional(name, npoints, deriv)¶
- build_b97d3bj_superfunctional(name, npoints, deriv)¶
- build_b97d3zero_superfunctional(name, npoints, deriv)¶
- build_blypd1_superfunctional(name, npoints, deriv)¶
- build_blypd2gr_superfunctional(name, npoints, deriv)¶
- build_blypd2p4_superfunctional(name, npoints, deriv)¶
- build_blypd3bj_superfunctional(name, npoints, deriv)¶
- build_blypd3zero_superfunctional(name, npoints, deriv)¶
- build_bp86d1_superfunctional(name, npoints, deriv)¶
- build_bp86d2gr_superfunctional(name, npoints, deriv)¶
- build_bp86d2p4_superfunctional(name, npoints, deriv)¶
- build_bp86d3bj_superfunctional(name, npoints, deriv)¶
- build_bp86d3zero_superfunctional(name, npoints, deriv)¶
- build_hcth120d3bj_superfunctional(name, npoints, deriv)¶
- build_hcth120d3zero_superfunctional(name, npoints, deriv)¶
- build_m05_2xd3zero_superfunctional(name, npoints, deriv)¶
- build_m05d3zero_superfunctional(name, npoints, deriv)¶
- build_pbe0d2gr_superfunctional(name, npoints, deriv)¶
- build_pbe0d2p4_superfunctional(name, npoints, deriv)¶
- build_pbe0d3bj_superfunctional(name, npoints, deriv)¶
- build_pbe0d3zero_superfunctional(name, npoints, deriv)¶
- build_pbed1_superfunctional(name, npoints, deriv)¶
- build_pbed2gr_superfunctional(name, npoints, deriv)¶
- build_pbed2p4_superfunctional(name, npoints, deriv)¶
- build_pbed3bj_superfunctional(name, npoints, deriv)¶
- build_pbed3zero_superfunctional(name, npoints, deriv)¶
text¶
- class Table(rows=(), row_label_width=10, row_label_precision=4, cols=(), width=16, precision=10)[source]¶
Class defining a flexible Table object for storing data.
Function to print text to output file in a banner of minimum width width and minimum three-line height for type = 1 or one-line height for type = 2.
util¶
Module with utility functions for use in input files.
- compare_integers(expected, computed, label)[source]¶
Function to compare two integers. Prints util.success() when value computed matches value expected. Performs a system exit on failure. Used in input files in the test suite.
- compare_matrices(expected, computed, digits, label)[source]¶
Function to compare two matrices. Prints util.success() when elements of matrix computed match elements of matrix expected to number of digits. Performs a system exit on failure to match symmetry structure, dimensions, or element values. Used in input files in the test suite.
- compare_strings(expected, computed, label)[source]¶
Function to compare two strings. Prints util.success() when string computed exactly matches string expected. Performs a system exit on failure. Used in input files in the test suite.
- compare_values(expected, computed, digits, label)[source]¶
Function to compare two values. Prints util.success() when value computed matches value expected to number of digits. Performs a system exit on failure. Used in input files in the test suite.
- compare_vectors(expected, computed, digits, label)[source]¶
Function to compare two vectors. Prints util.success() when elements of vector computed match elements of vector expected to number of digits. Performs a system exit on failure to match symmetry structure, dimension, or element values. Used in input files in the test suite.
- copy_file_from_scratch(filename, prefix, namespace, unit, move=False)[source]¶
Function to move file out of scratch with correct naming convention.
Arguments:
@arg filename full path to target file @arg prefix computation prefix, usually ‘psi’ @arg namespace context namespace, usually molecule name @arg unit unit number, e.g. 32 @arg move copy or move? (default copy)
Example:
Assume PID is 12345 and SCRATCH is /scratch/parrish/
- copy_file_to_scratch(‘temp’, ‘psi’, ‘h2o’, 32):
- -cp /scratch/parrish/psi.12345.h2o.32 .temp
- copy_file_to_scratch(‘/tmp/temp’, ‘psi’, ‘h2o’, 32):
- -cp /scratch/parrish/psi.12345.h2o.32 /tmp/temp
- copy_file_to_scratch(‘/tmp/temp’, ‘psi’, ‘’, 32):
- -cp /scratch/parrish/psi.12345.32 /tmp/temp
- copy_file_to_scratch(‘/tmp/temp’, ‘psi’, ‘’, 32, True):
- -mv /scratch/parrish/psi.12345.32 /tmp/temp
- copy_file_to_scratch(filename, prefix, namespace, unit, move=False)[source]¶
Function to move file into scratch with correct naming convention.
Arguments:
@arg filename full path to file @arg prefix computation prefix, usually ‘psi’ @arg namespace context namespace, usually molecule name @arg unit unit number, e.g. 32 @arg move copy or move? (default copy)
Example:
Assume PID is 12345 and SCRATCH is /scratch/parrish/
- copy_file_to_scratch(‘temp’, ‘psi’, ‘h2o’, 32):
- -cp ./temp /scratch/parrish/psi.12345.h2o.32
- copy_file_to_scratch(‘/tmp/temp’, ‘psi’, ‘h2o’, 32):
- -cp /tmp/temp /scratch/parrish/psi.12345.h2o.32
- copy_file_to_scratch(‘/tmp/temp’, ‘psi’, ‘’, 32):
- -cp /tmp/temp /scratch/parrish/psi.12345.32
- copy_file_to_scratch(‘/tmp/temp’, ‘psi’, ‘’, 32, True):
- -mv /tmp/temp /scratch/parrish/psi.12345.32
- success(label)[source]¶
Function to print a ‘label...PASSED’ line to screen. Used by util.compare_values() family when functions pass.
basislistdunning¶
Module (auto-generated from make_dunning.pl script) with commands building BasisFamily objects that encode the Dunning basis set orbital definitions in psi4/lib/basis/NOTES and fitting bases designed for those orbital bases.
aliases¶
Module with functions that call upon those in modules proc, driver, and wrappers.
- Place in this file quickly defined procedures such as
- aliases for complex methods
- simple modifications to existing methods
- run_mp2_5(name, **kwargs)[source]¶
Function that computes MP2.5 energy from results of a DETCI MP3 calculation.
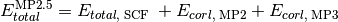
Psi variables: >>> energy('mp2.5')
- sherrillgroup_gold_standard(name='conv-mp2', **kwargs)[source]¶
Function to call the quantum chemical method known as ‘Gold Standard’ in the Sherrill group. Uses complete_basis_set() to evaluateo the following expression. Two-point extrapolation of the correlation energy performed according to corl_xtpl_helgaker_2().
![E_{total}^{\text{Au\_std}} = E_{total,\; \text{SCF}}^{\text{aug-cc-pVQZ}} \; + E_{corl,\; \text{MP2}}^{\text{aug-cc-pV[TQ]Z}} \; + \delta_{\text{MP2}}^{\text{CCSD(T)}}\big\vert_{\text{aug-cc-pVTZ}}](_images/math/4633e65e7989d388aa3eaf4c2a0163a8603d06db.png)
>>> energy('sherrillgroup_gold_standard')
qcdb¶
Module to facilitate quantum chemical computations on chemical databases. Contains Molecule class and physical constants from psi4 suite.
Module to largely replicate in python the psi4 libmints CoordValue and CoordEntry classes, which were developed by Justin M. Turney, with incremental improvements by other psi4 developers.
- class CartesianEntry(entry_number, Z, charge, mass, symbol, label, x, y, z)[source]¶
Class to hold all information about an atom, including its coordinate specification as three Cartesians.
- compute()[source]¶
Computes the values of the coordinates (in whichever units were inputted), returning them in a Vector
- class CoordEntry(entry_number, Z, charge, mass, symbol, label='')[source]¶
Class to
- class CoordValue(fixed=False, computed=False)[source]¶
An abstract class to handle storage of Cartesian coordinate values, which may be defined in terms of other variables through this mechanism, greatly simplifying Z-matrix specification, for example.
- class NumberValue(value, fixed=False)[source]¶
Specialization of CoordValue that is simply a number to be stored.
- class VariableValue(name, geometryVariables, negate=False, fixed=False)[source]¶
Specialization of CoordValue, where the current value depends on the list of geometry values stored by the molecule.
- class ZMatrixEntry(entry_number, Z, charge, mass, symbol, label, rto=None, rval=0, ato=None, aval=0, dto=None, dval=0)[source]¶
Class to hold all information about an atom, including its coordinate specification as any position of ZMatrix.
- class LibmintsMolecule(psi4molstr=None)[source]¶
Class to store the elements, coordinates, fragmentation pattern, charge, multiplicity of a molecule. Largely replicates psi4’s libmints Molecule class, developed by Justin M. Turney with incremental improvements by other psi4 developers. Major differences from the C++ class are: no basisset handling, no symmetry, no pubchem. This class translated so that databases can function independently of psi4.
>>> H2OH2O = qcdb.Molecule(""" 0 1 O1 -1.551007 -0.114520 0.000000 H1 -1.934259 0.762503 0.000000 H2 -0.599677 0.040712 0.000000 -- 0 1 X 0.000000 0.000000 0.000000 O2 1.350625 0.111469 0.000000 H3 1.680398 -0.373741 -0.758561 H4 1.680398 -0.373741 0.758561 no_com no_reorient units angstrom """)
>>> H2O = qcdb.Molecule.init_with_xyz('h2o.xyz')
- FullPointGroupList = ['ATOM', 'C_inf_v', 'D_inf_h', 'C1', 'Cs', 'Ci', 'Cn', 'Cnv', 'Cnh', 'Sn', 'Dn', 'Dnd', 'Dnh', 'Td', 'Oh', 'Ih']¶
- add_atom(Z, x, y, z, label='', mass=0.0, charge=0.0, lineno=-1)[source]¶
Add an atom to the molecule Z atomic number x cartesian coordinate y cartesian coordinate z cartesian coordinate symb atomic symbol to use mass mass to use if non standard charge charge to use if non standard lineno line number when taken from a string
- atom_at_position(b, tol=0.05)[source]¶
Tests to see of an atom is at the passed position b in Bohr with a tolerance tol.
>>> print H2OH2O.atom_at_position([1.35*(1.0/psi_bohr2angstroms), 0.10*(1.0/psi_bohr2angstroms), 0.0*(1.0/psi_bohr2angstroms)]) 3
- atom_to_unique(iatom)[source]¶
NYI Converts an atom number to the number of its generating unique atom. The return value is in [0, nunique).
- atom_to_unique_offset(iatom)[source]¶
NYI Converts an atom number to the offset of this atom in the list of generated atoms. The unique atom itself is allowed offset 0.
- center_of_mass()[source]¶
Computes center of mass of molecule (does not translate molecule).
>>> H2OH2O.center_of_mass() [-0.12442647346606871, 0.00038657002584110707, 0.0]
- charge(atom)[source]¶
Returns charge of atom (0-indexed). Related to SAD guess in libmints version.
>>> print H2OH2O.charge(4) 1.0
- charge_specified()[source]¶
Whether the charge was given by the user
>>> print H2OH2O.charge_specified() True
- create_molecule_from_string(text)[source]¶
Given a string geom of psi4-style geometry specification (including newlines to separate lines), builds a new molecule. Called from constructor.
- distance_matrix()[source]¶
Computes a matrix depicting distances between atoms. Prints formatted and returns array.
>>> H2OH2O.distance_matrix() Interatomic Distances (Angstroms) [1] [2] [3] [4] [5] [6] [1] 0.00000 [2] 0.95711 0.00000 [3] 0.96391 1.51726 0.00000 [4] 2.91042 3.34878 1.95159 0.00000 [5] 3.32935 3.86422 2.43843 0.95895 0.00000 [6] 3.32935 3.86422 2.43843 0.95895 1.51712 0.00000
- extract_fragments(reals, ghosts=[])[source]¶
Makes a copy of the molecule, returning a new molecule with only certain fragment atoms present as either ghost or real atoms reals: The list or int of fragments (1-indexed) that should be present in the molecule as real atoms. ghosts: The list or int of fragments (1-indexed) that should be present in the molecule as ghosts. (method name in libmints is extract_subsets. This is different in qcdb because the psi4 input parser tries to process lines with that term, giving rise to Boost:Python type conlicts.) See usage at extract_fragments().
- extract_subsets(reals, ghosts=[])[source]¶
Wrapper for extract_fragments(). See note there. This function can be used as long as not in psi4 input file. Use extract_fragments directly, then.
>>> H2OH2O.extract_subsets(2) # monomer B, unCP-corrected >>> H2OH2O.extract_subsets(2,1) # monomer B, CP-corrected >>> obj.extract_subsets(1,[2,3]) # monomer A, CP-corrected if obj is tri-molecular complex
- fcharge(atom)[source]¶
Returns charge of atom (0-indexed, includes dummies). Related to SAD guess in libmints version.
>>> print H2OH2O.fcharge(4) 8.0
- find_point_group(tol=1e-08)[source]¶
NYI Find computational molecular point group, user can override this with the “symmetry” keyword
- fix_orientation(_fix=True)[source]¶
Fix the orientation at its current frame (method name in libmints is set_orientation_fixed)
- flabel(atom)[source]¶
Returns the original label of the atom (0-indexed) as given in the input file (C2, H4) (includes dummies)
>>> print H2OH2O.flabel(4) O2
- fmass(atom)[source]¶
Returns mass of atom (0-indexed, includes dummies)
>>> print H2OH2O.fmass(4) 15.9949146196
- form_symmetry_information(tol=1e-08)[source]¶
NYI Initialize molecular specific symmetry information. Uses the point group object obtain by calling point_group()
- fsymbol(atom)[source]¶
Returns the cleaned up label of the atom (C2 => C, H4 = H) (includes dummies)
>>> print H2OH2O.fsymbol(4) O
- full_geometry()[source]¶
Returns the full (dummies included) geometry in Bohr as a N X 3 array.
>>> print H2OH2O.full_geometry() [[-2.930978460188563, -0.21641143673806384, 0.0], [-3.655219780069251, 1.4409218455037016, 0.0], [-1.1332252981904638, 0.0769345303220403, 0.0], [0.0, 0.0, 0.0], [2.5523113582286716, 0.21064588230662976, 0.0], [3.175492014248769, -0.7062681346308132, -1.4334725450878665], [3.175492014248769, -0.7062681346308132, 1.4334725450878665]]
- full_pg_n()[source]¶
NYI Return n in Cnv, etc.; If there is no n (e.g. Td) it’s the highest-order rotation axis.
- fx(atom)[source]¶
x position of atom (0-indexed, includes dummies) in Bohr
>>> print H2OH2O.fx(4) 2.55231135823
- fxyz(atom)[source]¶
Returns a Vector3 with x, y, z position of atom (0-indexed) in Bohr (includes dummies)
>>> print H2OH2O.fxyz(4) [2.5523113582286716, 0.21064588230662976, 0.0]
- fy(atom)[source]¶
y position of atom (0-indexed, includes dummies) in Bohr
>>> print H2OH2O.fy(4) 0.210645882307
- fz(atom)[source]¶
z position of atom (0-indexed, includes dummies) in Bohr
>>> print H2OH2O.fz(4) 0.0
- geometry()[source]¶
Returns the geometry in Bohr as a N X 3 array.
>>> print H2OH2O.geometry() [[-2.930978460188563, -0.21641143673806384, 0.0], [-3.655219780069251, 1.4409218455037016, 0.0], [-1.1332252981904638, 0.0769345303220403, 0.0], [2.5523113582286716, 0.21064588230662976, 0.0], [3.175492014248769, -0.7062681346308132, -1.4334725450878665], [3.175492014248769, -0.7062681346308132, 1.4334725450878665]]
- get_anchor_atom(vstr, line)[source]¶
Attempts to interpret a string vstr as an atom specifier in a zmatrix. Takes the current line for error message printing. Returns the atom number (adjusted to zero-based counting).
- get_coord_value(vstr)[source]¶
Attempts to interpret a string as a double, if not it assumes it’s a variable.
- get_full_point_group()[source]¶
NYI Return point group name such as C3v or S8. (method name in libmints is full_point_group)
- get_variable(vstr)[source]¶
Checks to see if the variable str is in the list, sets it to val and returns true if it is, and returns false if not.
- has_symmetry_element(op, tol=1e-08)[source]¶
NYI Whether molecule satisfies the vector symmetry operation op
- inertia_tensor()[source]¶
Compute inertia tensor.
>>> print H2OH2O.inertia_tensor() [[8.704574864178731, -8.828375721817082, 0.0], [-8.828375721817082, 280.82861714077666, 0.0], [0.0, 0.0, 281.249500988553]]
- init_with_checkpoint(chkpt)[source]¶
NYI Pull information from the chkpt object passed (method name in libmints is init_with_chkpt)
- init_with_io(psio)[source]¶
NYI Pull information from a chkpt object created from psio (method name in libmints is init_with_psio)
- classmethod init_with_xyz(xyzfilename)[source]¶
Pull information from an XYZ file. No fragment or chg/mult info detected.
>>> H2O = qcdb.Molecule.init_with_xyz('h2o.xyz')
- is_axis(origin, axis, order, tol=1e-08)[source]¶
Is axis an axis of order order with respect to origin?
- is_linear_planar(tol=1e-08)[source]¶
Is the molecule linear, or planar?
>>> print H2OH2O.is_linear_planar() (False, False)
- is_variable(vstr)[source]¶
Checks to see if the variable str is in the list, returns true if it is, and returns false if not.
>>> H2OH2O.is_variable('R') False
- label(atom)[source]¶
Returns the original label of the atom (0-indexed) as given in the input file (C2, H4).
>>> print H2OH2O.label(4) H3
- static like_world_axis(axis, worldxaxis, worldyaxis, worldzaxis)[source]¶
Returns which worldaxis axis most overlaps with. Inverts axis when indicated.
- multiplicity_specified()[source]¶
Whether the multiplicity was given by the user
>>> print H2OH2O.multiplicity_specified() True
- nactive_fragments()[source]¶
The number of active fragments in the molecule.
>>> print H2OH2O.nactive_fragments() 2
- nfrozen_core(depth='SMALL')[source]¶
Number of frozen core for molecule given freezing state.
>>> print H2OH2O.nfrozen_core() 2
- nuclear_repulsion_energy()[source]¶
Computes nuclear repulsion energy.
>>> print H2OH2O.nuclear_repulsion_energy() 36.6628478528
- nuclear_repulsion_energy_deriv1()[source]¶
Computes nuclear repulsion energy derivatives
>>> print H2OH2O.nuclear_repulsion_energy_deriv1() [[3.9020946901323774, 2.76201566471991, 0.0], [1.3172905807089021, -2.3486366050337293, 0.0], [-1.8107598525022435, -0.32511212499256564, 0.0], [-1.217656141385739, -2.6120090867576717, 0.0], [-1.0954846384766488, 1.2618710760320282, 2.1130743287465603], [-1.0954846384766488, 1.2618710760320282, -2.1130743287465603]]
- orientation_fixed()[source]¶
Get whether or not orientation is fixed.
>>> H2OH2O.orientation_fixed() True
- print_bond_angles()[source]¶
Print the geometrical parameters (bond_angles) of the molecule.
>>> print H2OH2O.print_bond_angles() Bond Angles (degrees) Angle 2-1-3: 104.337 Angle 2-1-4: 109.152 Angle 2-1-5: 117.387 ...
- print_dihedrals()[source]¶
Print the geometrical parameters (dihedrals) of the molecule.
>>> print H2OH2O.print_dihedrals() Dihedral Angles (Degrees) Dihedral 1-2-3-4: 180.000 Dihedral 1-2-3-5: 133.511 Dihedral 1-2-3-6: 133.511 ...
- print_distances()[source]¶
Print the geometrical parameters (distances) of the molecule. suspect libmints version actually prints Bohr.
>>> print H2OH2O.print_distances() Interatomic Distances (Angstroms) Distance 1 to 2 0.957 Distance 1 to 3 0.964 Distance 1 to 4 2.910 ...
- print_full()[source]¶
Print full atom list. Same as print_out() only displays dummy atoms.
- print_in_input_format()[source]¶
Print the molecule in the same format that the user provided. Only returns if Zmat or variable input.
- print_out()[source]¶
Print the molecule. (method name in libmints is print)
>>> H2OH2O.print_out() Geometry (in Angstrom), charge = -2, multiplicity = 3: Center X Y Z ------------ ----------------- ----------------- ----------------- O -1.551007000000 -0.114520000000 0.000000000000 H -1.934259000000 0.762503000000 0.000000000000 H -0.599677000000 0.040712000000 0.000000000000 O 1.350625000000 0.111469000000 0.000000000000 H 1.680398000000 -0.373741000000 -0.758561000000 H 1.680398000000 -0.373741000000 0.758561000000
- print_out_in_angstrom()[source]¶
Print the molecule in Angstroms. Same as print_out() only always in Angstroms. (method name in libmints is print_in_angstrom)
- print_out_in_bohr()[source]¶
Print the molecule in Bohr. Same as print_out() only in Bohr. (method name in libmints is print_in_bohr)
- print_out_of_planes()[source]¶
Print the geometrical parameters (out_of_planes) of the molecule.
>>> print H2OH2O.print_out_of_planes() Out-Of-Plane Angles (Degrees) Out-of-plane 1-2-3-4: 0.000 Out-of-plane 1-2-3-5: -7.373 Out-of-plane 1-2-3-6: 7.373 ...
- reinterpret_coordentries()[source]¶
Reinterpret the fragments for reals/ghosts and build the atom list.
- reinterpret_coordentry(rc)[source]¶
Do we reinterpret coordentries during a call to update_geometry? (method name in libmints is set_reinterpret_coordentry)
- rotate(R)[source]¶
Rotates the molecule using rotation matrix R.
>>> H2OH2O.rotate([[0,-1,0],[-1,0,0],[0,0,1]])
- rotate_full(R)[source]¶
Rotates the full molecule using rotation matrix R.
>>> H2OH2O.rotate_full([[0,-1,0],[-1,0,0],[0,0,1]])
- rotational_constants(tol=1e-08)[source]¶
Compute the rotational constants and return them in wavenumbers
- save_string_xyz()[source]¶
Save a string for a XYZ-style file.
>>> H2OH2O.save_string_xyz() 6 -2 3 water_dimer O -1.551007000000 -0.114520000000 0.000000000000 H -1.934259000000 0.762503000000 0.000000000000 H -0.599677000000 0.040712000000 0.000000000000 O 1.350625000000 0.111469000000 0.000000000000 H 1.680398000000 -0.373741000000 -0.758561000000 H 1.680398000000 -0.373741000000 0.758561000000
- save_to_checkpoint(chkpt, prefix='')[source]¶
NYI Save information to checkpoint file (method name in libmints is save_to_chkpt)
- set_basis_by_label(label, name, type='BASIS')[source]¶
NYI Assigns basis name to all atoms with label.
- set_basis_by_number(number, name, type='BASIS')[source]¶
NYI Assigns basis name to atom number number (1-indexed, includes dummies).
- set_basis_by_symbol(symbol, name, type='BASIS')[source]¶
NYI Assigns basis name to all symbol atoms.
- set_com_fixed(_fix=True)[source]¶
NYI Fix the center of mass at its current frame. Not used in libmints so not implemented.
- set_full_geometry(geom)[source]¶
Sets the full geometry (dummies included), given a N X 3 array of coordinates geom in Bohr.
>>> H2OH2O.set_full geometry([[1,2,3],[4,5,6],[7,8,9],[0,0,0],[-1,-2,-3],[-4,-5,-6],[-7,-8,-9]])
- set_geometry(geom)[source]¶
Sets the geometry, given a N X 3 array of coordinates geom in Bohr.
>>> H2OH2O.set_geometry([[1,2,3],[4,5,6],[7,8,9],[-1,-2,-3],[-4,-5,-6],[-7,-8,-9]])
- set_multiplicity(mult)[source]¶
Sets the multiplicity (defined as 2Ms + 1)
>>> H2OH2O.set_multiplicity(3)
- set_variable(vstr, val)[source]¶
Assigns the value val to the variable labelled string in the list of geometry variables. Also calls update_geometry()
- symbol(atom)[source]¶
Returns the cleaned up label of the atom (C2 => C, H4 = H) (0-indexed)
>>> print H2OH2O.symbol(4) H
- symmetrize()[source]¶
NYI Force the molecule to have the symmetry specified in pg. This is to handle noise coming in from optking.
- symmetry_frame(tol=1e-08)[source]¶
Determine symmetry reference frame. If noreorient is not set, this is the rotation matrix applied to the geometry in update_geometry.
>>> print H2OH2O.symmetry_frame() [[1.0, -0.0, 0.0], [0.0, 1.0, 0.0], [0.0, -0.0, 1.0]]
- symmetry_from_input()[source]¶
Returns the symmetry specified in the input.
>>> print H2OH2O.symmetry_from_input() C1
- update_geometry()[source]¶
Updates the geometry, by (re)interpreting the string used to create the molecule, and the current values of the variables. The atoms list is cleared, and then rebuilt by this routine. This function must be called after first instantiation of Molecule.
>>> H2 = qcdb.Molecule("H\nH 1 0.74\n") >>> print H2.natom() 0 >>> H2.update_geometry() >>> print H2.natom() 2
Module to facilitate quantum chemical computations on chemical databases. Contains Molecule class and physical constants from psi4 suite.
File to
Module with non-generic exceptions classes.
- exception FeatureNotImplemented(msg)[source]¶
Error called for functions defined but not yet implemented. Also for functions defined that will never be implemented.
- exception IncompleteAtomError(msg)[source]¶
Error raised when not all variables in an atom specification have been defined at compute time. May be a temporary situation so message not printed but appears as traceback when error persists.
- exception ValidationError(msg)[source]¶
Error called for problems with syntax input file. Prints error message msg to standard output stream.
- class Molecule(psi4molstr=None)[source]¶
Class to store python extensions to the MoleculeLibmints class. Multiple classes allows separation of libmints and extension methods.
- BFS()[source]¶
Perform a breadth-first search (BFS) on the real atoms in molecule, returning an array of atom indices of fragments. Relies upon van der Waals radii and so faulty for close (esp. hydrogen-bonded) fragments. Original code from Michael S. Marshall.
- auto_fragments()[source]¶
Detects fragments in an unfragmented molecule using BFS algorithm. Returns a new Molecule in Cartesian, fixed-geom (no variable values), no dummy-atom format. Any non-default charge and multiplicity assigned to first fragment.
- classmethod init_with_xyz(xyzfilename, no_com=False, no_reorient=False)[source]¶
Pull information from an XYZ file. No fragment info detected. Charge, multiplicity, tagline pulled from second line if available.
>>> H2O = qcdb.Molecule.init_with_xyz('h2o.xyz')
- save_string_for_psi4()[source]¶
Returns a string of Molecule formatted for psi4. Includes fragments and reorienting, if specified.
>>> print H2OH2O.save_string_for_psi4() 6 0 1 O -1.55100700 -0.11452000 0.00000000 H -1.93425900 0.76250300 0.00000000 H -0.59967700 0.04071200 0.00000000 -- 0 1 @X 0.00000000 0.00000000 0.00000000 O 1.35062500 0.11146900 0.00000000 H 1.68039800 -0.37374100 -0.75856100 H 1.68039800 -0.37374100 0.75856100 units Angstrom
Elemental masses (most common isotope), symbols, and atomic numbers from psi4.
Stuff stolen from psi. Should import or not as necessary or some better way. Apologies to the coders.
- compare_matrices(expected, computed, digits, label)[source]¶
Function to compare two matrices. Prints util.success() when elements of matrix computed match elements of matrix expected to number of digits. Performs a system exit on failure to match symmetry structure, dimensions, or element values. Used in input files in the test suite.
- compare_values(expected, computed, digits, label)[source]¶
Function to compare two values. Prints util.success() when value computed matches value expected to number of digits. Performs a system exit on failure. Used in input files in the test suite.
- query_yes_no(question, default=True)[source]¶
Ask a yes/no question via raw_input() and return their answer.
question is a string that is presented to the user. default is the presumed answer if the user just hits <Enter>. It must be yes (the default), no or None (meaning an answer is required of the user).
The return value is one of True or False.
File for accessory procedures in the chem module. Credit for the libmints vector3 class to Justin M. Turney and incremental improvements by other psi4 developers.